DOI:
10.1039/C6RA13669A
(Paper)
RSC Adv., 2016,
6, 80485-80492
Improvement of interfacial strength and thermal stability of carbon fiber composites by directly grafting unique particles: functionalized mesoporous silicas
Received
26th May 2016
, Accepted 29th July 2016
First published on 1st August 2016
Abstract
A facile process was used to introduce vinyl functionalized mesoporous silicas onto a carbon fiber to improve the interfacial strength of composites. The characterization of successfully grafted vinyl groups on mesoporous silicas was determined by Fourier transform infrared (FTIR) and X-ray diffraction (XRD). The morphology of the vinyl functionalized mesoporous silicas uniformly distributed on the surface of carbon fiber was characterized by scanning electron microscopy (SEM), which indicated that the homogeneous dispersion of vinyl functionalized mesoporous silicas on a fiber may benefit the interfacial adhesion of matrix. Interfacial shear strength and thermal stability of the composites were improved by increasing the vinyl functionalized mesoporous silicas. This effect arose from the fact that functionalized mesoporous silicas serve as a “rivet joint” produced from chemical bonding and physical interlocking among the mesoporous silicas, carbon fiber and matrix. Notably, introduction of functional mesoporous silicas may be applied for fabricating high performance, multifunctional carbon fiber composites.
1. Introduction
It is well-known that carbon fiber composites exhibit superior performance with high specific strength, high stiffness, high thermal stability and corrosion resistance, which correspond to great potential in the aviation and automotive industries.1 Most significantly, carbon fiber plays an irreplaceable role in automobile and aircraft lightening due to its high strength to weight ratio.2 Due to these excellent properties, numerous researchers have paid more attention to the combination of a carbon fiber with a matrix. Thermosetting resins are frequently utilized as the matrix for carbon fiber composites.3 Vinyl ester resin is utilized as the matrix polymer of the carbon fiber composites due to its high mechanical property and good processability. Moreover, it exhibits superior properties such as excellent resistance to acids and bases, low curing shrinkage and low cost. Because of these excellent properties, carbon fiber/vinyl ester composites are considered as potential materials in industry and aviation.4
However, the adhesive property of thermosetting resins with carbon fiber is sorely in need of improvement due to the hydrophobic nature and inertia of a carbon fiber. It was well recognized that for carbon fiber reinforcing composites, the resulting mechanical properties mainly depend on the interfaces between the fibers and matrix.5–7 Many methods have been used to improve the surface adhesion property of carbon fiber.3,8,9 Electrochemical methods are widely used to oxidize the surface. Nevertheless, the waste electrolytic solution for this method does not conform to the highly desired environment protection and energy-saving requirements. Recently, nanoparticles, such as nano-ZnO, SiO2 and graphene oxide, improved the mechanical properties of the composites and have been reported by many literature on account of the high specific surface area and quantum dimension effect.10–12 Direct grafting of CNTs onto a carbon fiber is identified as a promising method that can simultaneously provide stronger interfacial bonding and matrix strength,13,14 The key challenge in this method is the homogeneous distribution of CNTs on the fiber.15 Nanoscale mesoporous silica is a rattling candidate for the inorganic component of composites due to its excellent properties, which plays an important role in catalysis,16 adsorption,17 and drug delivery.18,19 Furthermore, the mesoporous structure can be regarded as a nanoreactor due to its particular structure, and the reaction will react in nanohole of mesoporous silica which named in situ polymerization.20–23 Mesoporous materials, MCM-41, has been used as a reinforcing material to enhance the mechanical and thermal properties of the polymer material.24,25 Moreover, MCM-41 is less expensive and contains abundant silanol groups, which can facilitate its post-functionalization.
In this study, mesoporous materials (MCM-41) were introduced onto the carbon fiber to increase roughness and chemical bonding of the carbon fiber to enhance the interfacial strength between resins and fibers. Vinyl groups were grafted onto MCM-41 for reinforcing the interaction between MCM-41 and vinyl-resin via radical polymerization with a hot-pressing process. An easy-to-use method was used to graft functionalized mesoporous silicas particles onto CF by a covalent ester linkage at low temperature and without other coupling agents. CF/VE composites with different contents of MCM-41 particles were prepared. Thermal stability of the composites was investigated. The relationship between composites properties and interfacial strength is discussed. The IFSS of V-MCM-41 grafted carbon fiber/vinyl resin composite increases by 62.3% compared with pristine VE/CF composites.
2. Experimental
2.1 Materials
Polyacrylonitrile (PAN)-based type carbon fiber was obtained from Toray Co, under the T300 trade name. Vinyl ester was purchased from Shanghai Fuchem Co. Nitric acid (HNO3, 68%), potassium bromide (KBr), cetyltrimethyl ammonium bromide (CTAB), tetraethylorthosilicate (TEOS), cumene hydroperoxide, acetone, ammonium hydroxide, alcohol, toluene, acetic acid, and sulfuric acid were provided from Aladdin Co, Shanghai China. Triethoxyvinylsilane (VTEO) was procured from Shanghai Macklin Biochemical Co.
2.2 Preparation of composites
2.3 Characterization
Fourier transform infrared spectroscopy (FTIR) was used to determine the functional groups of the samples within the 400–4000 cm−1 region (Bruker-IFS 66 V/S). Structures of the products were characterized by X-ray diffraction (XRD) using a Rigaku D/max 2550. The Brunauer–Emmett–Teller (BET) specific surface area was calculated using the Barrett–Joyner–Halenda (BJH) model. Scanning electron microscopy (SEM) was performed on JSM-6510 and JSM-5600 microscopes to characterize the microstructures of the fiber and the composites cross-sections. The XPS measurement of the fiber surface was performed using a VG Scientific ESCA LABMK-II spectrometer. The interface shear strength for a single fiber was carried out on a test machine (FA-620, East Rong Corporation of Japan) with a cross-head speed of 2 mm min−1. The interfacial strength, τ, between the CF and resin can be calculated as follows:26where F is the maximum pulling load of the CF and dcf and lcf are the diameter of CF and the embedded length of CF into the matrix, respectively. The glass transition behaviors of composites were determined using differential scanning calorimetry (NETZSCH DSC 200 F3). All experiments were performed in a nitrogen atmosphere at a heating rate of 10 °C min−1. The thermal stability of the composites was determined by a Metter Toledo TGA 2. The samples were heated from 30 to 900 °C at a heating rate of 10 °C min−1 in a nitrogen atmosphere.
3. Results and discussion
3.1 Characterization of MCM-41 and vinyl functionalized MCM-41
In this study, FTIR spectra were first obtained and used to identify the formations of MCM-41 and V-MCM-41 after synthesis. As shown in Fig. 4(a), the peaks at 456 cm−1, 798 cm−1 and 1080 cm−1, respectively, correspond to the absorption bands of MCM-41.27 While comparing the two curves, new obvious peaks appeared at 1412 and 1603 cm−1 and are shown in Fig. 4(b). The bands around 1603 cm−1 and 1412 cm−1 can be attributed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching mode and the CH2 in plane bending deformation for Si–CHCH2, respectively. The peak at 3000 cm−1 arises from the stretching vibration of C–H bonds due to terminal methylene (CH2) and alkyl substituted vinyl (Si–CHCH2).28 Clearly, the broad peak attributed to stretching vibration of –OH group at 3100 cm−1 slightly decreased in VTEO functionalized MCM-41. Thus, these data indicate that modifying with vinyl on MCM-41 was successfully performed.
C stretching mode and the CH2 in plane bending deformation for Si–CHCH2, respectively. The peak at 3000 cm−1 arises from the stretching vibration of C–H bonds due to terminal methylene (CH2) and alkyl substituted vinyl (Si–CHCH2).28 Clearly, the broad peak attributed to stretching vibration of –OH group at 3100 cm−1 slightly decreased in VTEO functionalized MCM-41. Thus, these data indicate that modifying with vinyl on MCM-41 was successfully performed.
 |
| | Fig. 4 FTIR spectra of MCM-41 and vinyl functionalized MCM-41. | |
To verify the nanoporous structure, small angle X-ray diffraction and N2 adsorption and desorption isotherms of functionalized MCM-41 were performed. The small angle XRD pattern is shown in Fig. 5. As previously reported, MCM-41 synthesized particles exhibited one intense (100) diffraction peak and two additional small reflections at (110) and (200), which correspond to the hexagonal structure.24 Curve b shows that the grafted mesoporous pore morphology is still the long-range ordered hexagonal mesoporous structure. According to the isotherms in Fig. 5(B), typical IV curves were exhibited, which are characteristic of uniform mesoporous silicas treatment.29 The pore size of MCM-41 was 2.63 nm, whereas the corresponding pore size of V-MCM-41 was 2.11 nm smaller than before, suggesting that triethoxyvinylsilane was successfully grafted into mesosilicas. Based on the abovementioned analyses, functionalized V-MCM-41 was successfully prepared but also had no damage to its special structure, which laid the foundation for further research.
 |
| | Fig. 5 (A) XRD pattern of MCM-41 before and after modification; (B) N2 adsorption and desorption isotherms. | |
3.2 Characteristics of V-MCM-41 grafted onto a carbon fiber
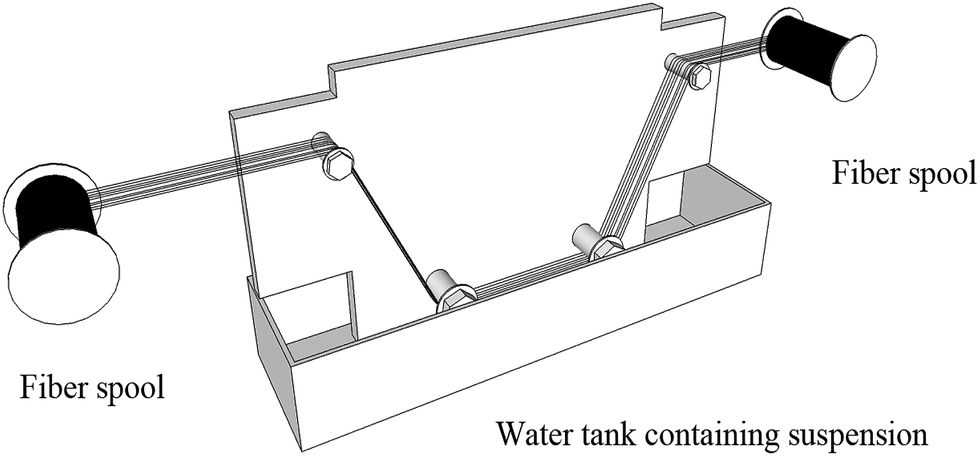
Vinyl functionalized MCM-41 particles were dispersed via a dipping method using a self-designed system. A V-MCM-41-incorporated CF was imaged by SEM to investigate the surface morphology. A representative image is shown in Fig. 6(a), which reveals a relatively smooth surface. Another similar surface pattern is shown in Fig. 6(b), which indicates that the 2.0 wt% V-MCM-41 was homogeneously grafted onto the CF slide surface. It is well known that the roughness of carbon fiber plays an important role in adhesion of the fiber and matrix.30 The V-MCM-41 particles grafted onto fiber like a “nail”, which mechanically interlocks making desquamating of the resin difficult from the carbon fiber surface. After treatment with nitric acid, some –COOH and –OH groups were produced on the carbon fiber,31 and chemical bonding may form with the Si–OH on mesoporous silicas. Therefore, it is reasonable to deduct that adhesion between fiber and matrix could be improved with increasing roughness of the V-MCM-41 grafted carbon fiber.
 |
| | Fig. 6 SEM micrographs of V-MCM-41 deposited onto a fiber surface: (a) COOH–CF without grafting (b) COOH–CF grafted with 2.0 wt% of V-MCM-41. | |
In order to prove our conjecture, FTIR was performed to reveal the reaction between CF and V-MCM-41. The spectra, as shown in Fig. 7, indicate the absence of hydroxyl –OH (3100–3300 cm−1) and carbonyl –CO groups (1540–1740 cm−1) in the carbon fiber after acid treatment.32 The peaks at 1218 and 1058 cm−1 are attributed to C–O band in alcohols and ethers.33 Notably, comparing with COOH–CF, a red shift was observed in the position of C–O (from 1058 to 1080 cm−1) for V-MCM-41–CF, which might be attributed to the formation of ester groups between CF and V-MCM-41.
 |
| | Fig. 7 FTIR spectra of COOH–CF and V-MCM-41–CF. | |
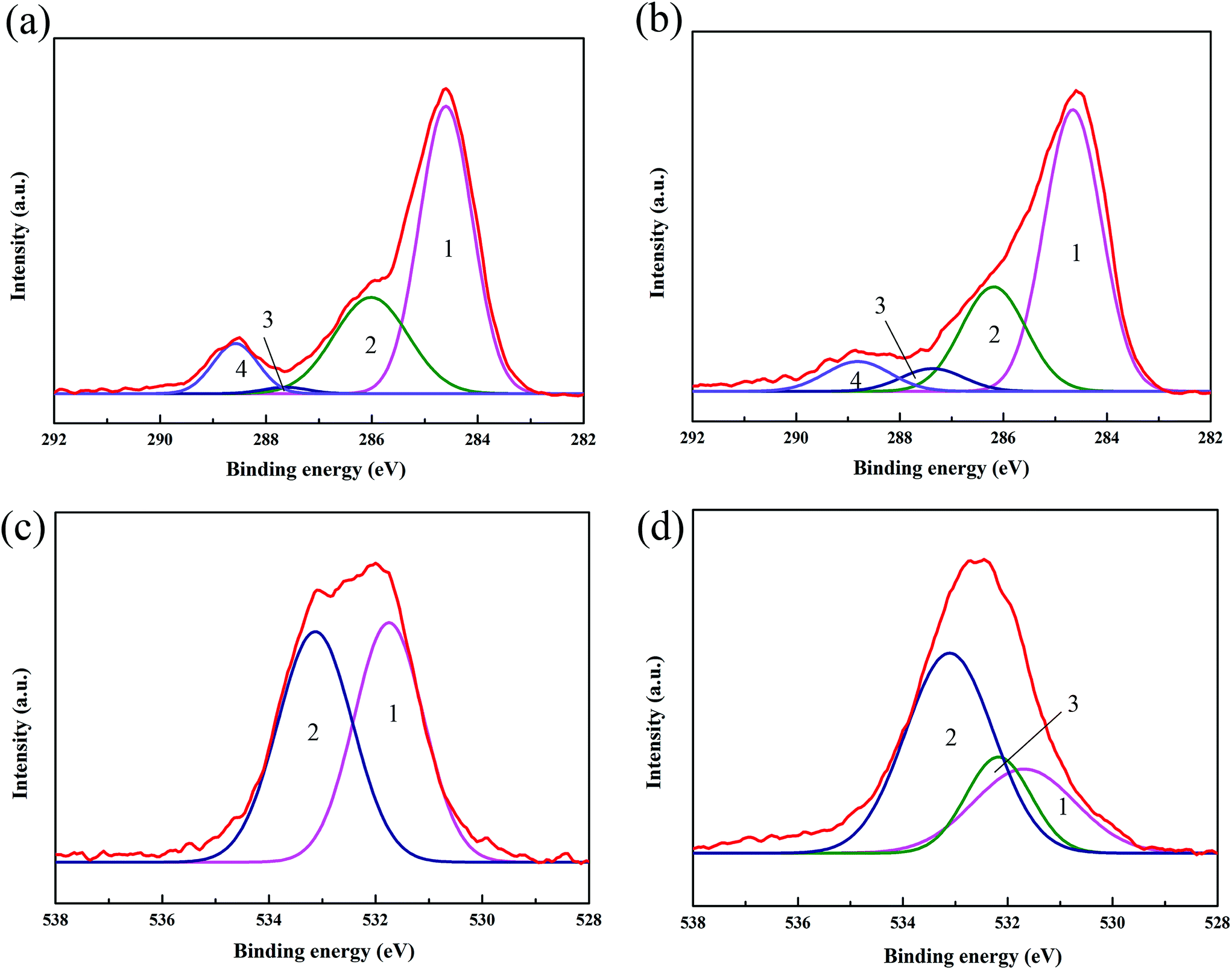
The interaction between CF and V-MCM-41 was further investigated using XPS. To determine differences in chemical composition for these samples, XPS C1s and O1s spectra were obtained and are shown in Fig. 8. The peaks of C1s (Fig. 8(a) and (b)) at 284.8 eV (peak 1) originate from the sp2 hybridized graphitic carbon; the peaks at 286.3 eV (peak 2), 287.8 eV (peak 3), and 288.8 eV (peak 4) can be attributed to C–O (alcohol, ether, phenol), CO (carbonyl), and –COOH (carboxylic) groups, respectively.34 It can be seen that the peak for the CO groups increased a lot and the peaks for –COOH groups significantly decreased, which could be ascribed to the formation of an ester linkage. To further evaluate the change in chemical bonds of the samples, the O1s peaks were analyzed and are shown in Fig. 8(c) and (d). The peaks at 531.1 eV (peak 1) and 533.1 eV (peak 2) originated from oxygen doubly bonded to carbon, respectively.35 However, it is obviously observed in Fig. 8(d) that V-MCM-41 grafted on CF samples produced an extra peak at 532.3 eV (peak 3), which originated from carbonyl oxygen atoms in ester groups, further explaining the formation of an ester linkage between the V-MCM-41 and carbon fiber.36
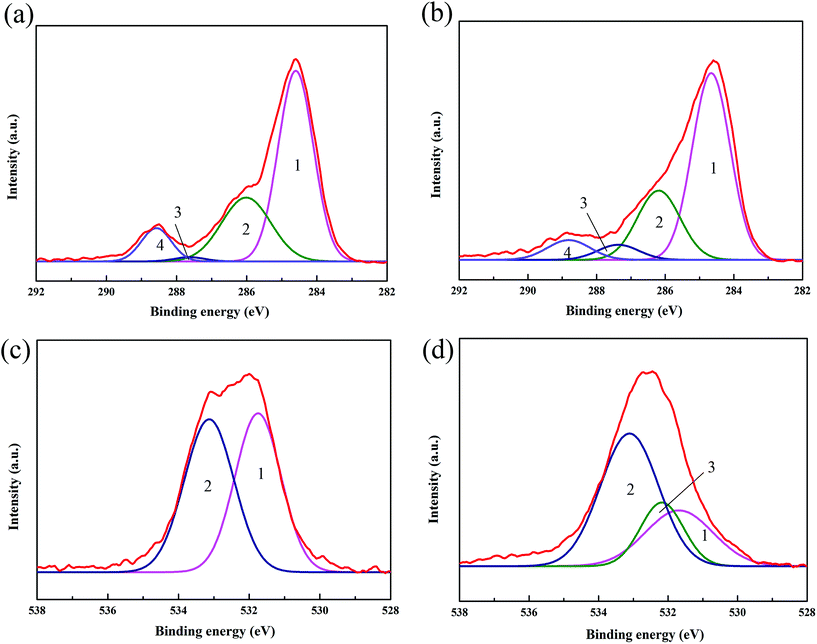 |
| | Fig. 8 XPS spectra of samples; curve fitting of C1s peak area to surface functional groups of (a) COOH–CF and (b) V-MCM-41 grafted on CF, curve fit of O1s photoelectron peaks of (c) COOH–CF and (d) V-MCM-41 grafted on CF. | |
3.3 Characteristics of VE/V-MCM-41/CF composites
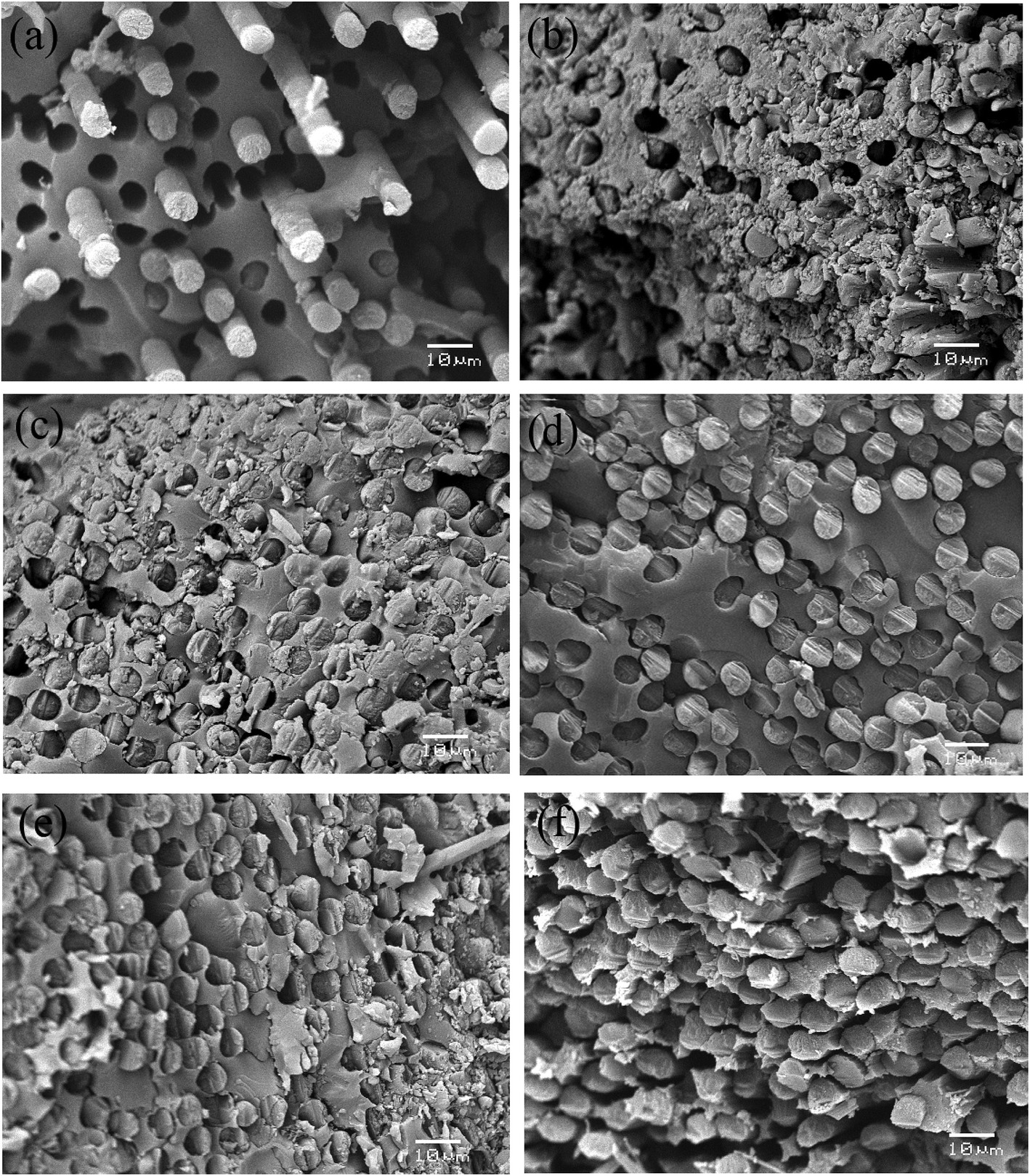
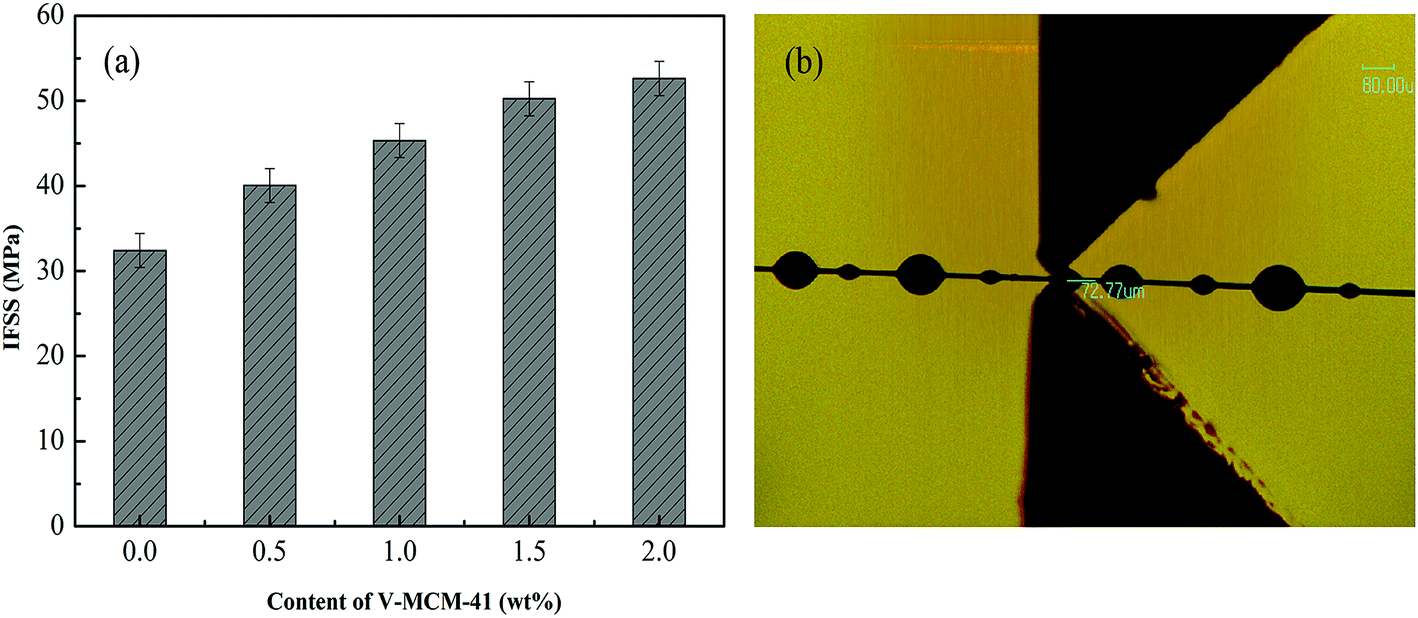
The morphology of cross-section is an effective assessment indicator for interfacial properties of composites. In order to access the fracture morphology of the composites, the cross-sections of the specimens, which were created by a brittle fracture, were examined. Fig. 9 presents typical SEM images of the composites before and after V-MCM-41 was treated. It is clearly shown in Fig. 9(a) that the composite which not treated with V-MCM-41 has a ragged fracture plane with many holes due to the carbon fiber pulling out from the matrix, and the fiber is exposed outward with no resin, indicating a poor combination of the carbon fiber and resin. In Fig. 9(b)–(e), with increasing V-MCM-41 content, few fibers pull out from the matrix and fewer holes arise. This phenomenon may be attributed two reasons: on one hand, the unsaturated carbon–carbon bonds of vinyl resin react with vinyl functionalized MCM-41 via free radical polymerization in consolidation process; on the other hand, molecular chains of the resin may embed in the channels of MCM-41 forming physical crosslinking sites, as shown in Fig. 1(b). The integration of them contributes to enhancing the surface roughness and strengthening the chemical bond, which consequently improves the interfacial bond strength by creating local interlocks between the fiber and resin. Therefore, depositing V-MCM-41 onto a carbon fiber is helpful to improve the interphase property of the composites.
 |
| | Fig. 9 Fracture morphologies of the composites: (a) COOH–CF without grafting, (b–e) grafted with 0.5, 1.0, 1.5, 2.0 wt% V-MCM-41 content, respectively, (f) COOH–CF grafted with 0.5 wt% content of untreated MCM-41. | |
The composites containing 0.5 wt% unmodified MCM-41 particles on the fiber were also manufactured in order to examine whether they would have a similar effect. As shown in Fig. 9(f), the fibers were pulled out from the matrix and the space between fiber and resin was very big, showing poor interfacial bonding for these composites.37 This may be attributed to the agglomeration of untreated particles leading to material imperfections.
After grafting V-MCM-41, the surface roughening and functional group bonding may play predominant roles in the interfacial properties, hence the microbond test was used to evaluate the interfacial properties. Single fiber composites were tested by FA-620, and the data are given in Fig. 10. As shown in Fig. 10(a), the IFSS of untreated fiber was 32.42 MPa, whereas the deposition of grafted V-MCM-41 gave rise to an apparent increase of IFSS for the composites. There were 23.5%, 40.0%, and 50.24% increase in the IFSS of the composites after 0.5, 1.0, 1.5 wt% V-MCM-41 grafting, respectively. A significant increase of IFSS was noted with 2.0 wt% V-MCM-41 grafted onto a fiber, as shown in Fig. 10. The influence of IFSS may contribute to the combination of chemical bonding and physical crosslinking.
 |
| | Fig. 10 (a) Interfacial shear strength of the composites, (b) optical microscope image of the microdroplet. | |
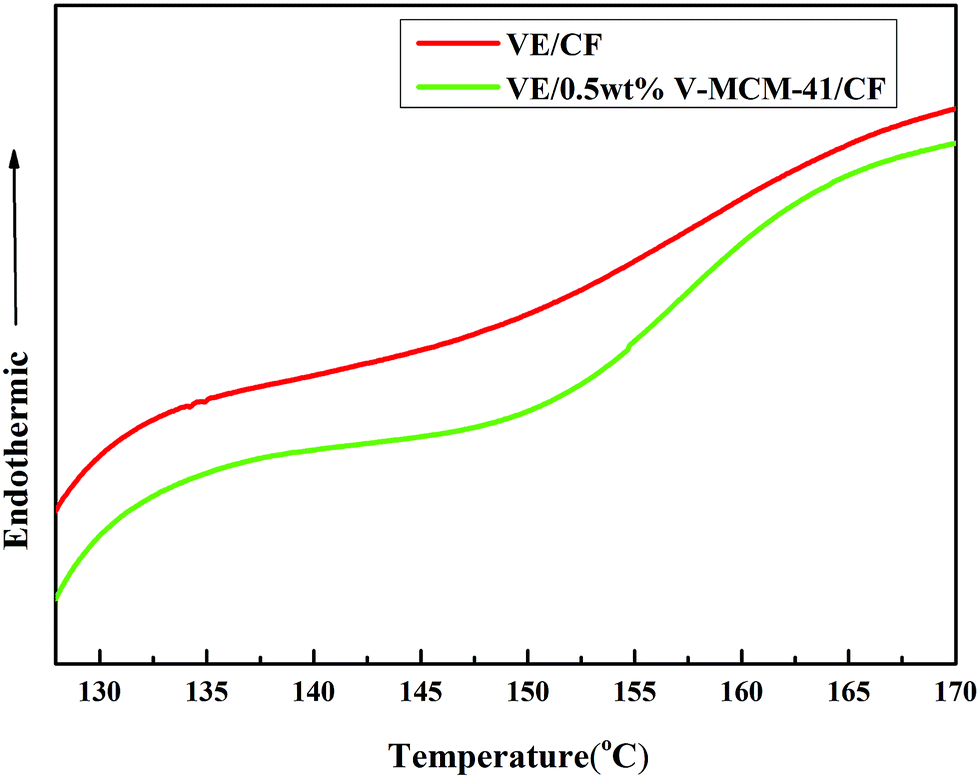
In order to explore the reaction of free radical polymerization in the consolidation process between unsaturated carbon–carbon bonds of vinyl resin and vinyl functionalized MCM-41, the glass transition behavior of neat VE/CF and VE/0.5 wt%V-MCM-41/CF composites, used immediately after preparation, was measured by DSC and are shown in Fig. 11. The Tg of the neat sample was about 149.30 °C, whereas the grafted 0.5 wt% vinyl MCM-41 composites showed a higher Tg of about 155.16 °C. The results indicate that incorporating V-MCM-41 into the VE matrix somewhat restricted the thermal motion of the polymer chains in the existing covalent bonds between VE matrix and vinyl functionalized MCM-41 (ref. 4).
 |
| | Fig. 11 DSC curves of neat VE/CF and VE/0.5 wt%V-MCM-41 composites. | |
Based on reports of other scholars, mesoporous materials may affect the thermal properties of polymers.27,38 TGA measurements were carried out to evaluate the effect of the V-MCM-41 on VE/CF composites aimed to confirm whether a similar trend existed, and the corresponding thermogravimetric curves are shown in Fig. 12. The epitaxial decomposition temperature (Tei) and the temperature at 10 wt% loss (T10) and 20 wt% loss (T20) for composite laminates, and the maximum decomposition temperature (Tmax) are tabulated in Table 1. It can be observed that the thermal stability of VE/CF laminates increased with the addition of V-MCM-41. The Tei for the VE/CF laminates was 381.57 °C, and the Tei for the VE/2.0 wt%V-MCM-41/CF was increased to 389.61 °C. The T10 and T20 increased from 380.83 °C to 399.00 °C and 398.50 °C to 422.00 °C, respectively. Moreover, Tmax was improved from 415.33 °C to 426.00 °C. All the data demonstrated that V-MCM-41 gives assistance to the thermostability of composites. The influence of thermal stability could be ascribed to strong interaction between the VE matrix and carbon fiber besides the special structure of V-MCM-41 hinders the segmental motion of polymer chain.39 In addition, long-range ordered hexagonal mesoporous structure of MCM-41 may block the diffusion of volatile gases, resulting in difficulty in contacting with air. The char yield of the composites increased with the V-MCM-41 weight percentage, probably because the resin chains on the V-MCM-41 surface and inside pore were more difficult to decompose than non-grafted V-MCM-41 composites. At same time, grafted particles on fiber may restrain the fiber from contacting with air.
 |
| | Fig. 12 TGA thermograms of the VE/V-MCM-41/CF laminates. | |
Table 1 Thermal properties of VE/V-MCM-41/CF laminatea
| Sample |
Tei (°C) |
Tmax (°C) |
T10 (°C) |
T20 (°C) |
| Tei and Tmax are the epitaxial decomposition temperature and maximum decomposition temperature, respectively. T10 and T20 correspond to the temperatures at 10 wt% and 20 wt% losses, respectively. |
| VE/CF |
381.57 |
415.33 |
380.83 |
398.50 |
| VE/0.5 wt%V-MCM-41/CF |
388.98 |
426.00 |
388.76 |
405.33 |
| VE/1.0 wt%V-MCM-41/CF |
390.20 |
426.50 |
390.33 |
415.67 |
| VE/1.5 wt%V-MCM-41/CF |
390.84 |
426.00 |
392.80 |
420.17 |
| VE/2.0 wt%V-MCM-41/CF |
389.61 |
426.00 |
399.00 |
422.00 |
4. Conclusion
In this study, mesoporous silica-MCM-41 was successfully functionalized with VTEO, generating V-MCM-41, and was then grafted onto a carbon fiber using a simple process. Compression mould forming was used to fabricate VE/V-MCM-41/CF composites with different V-MCM-41 content. Introduction of V-MCM-41 onto carbon fiber improved the interfacial properties of the composites, which was attributed to the special structure of mesoporous silicas. The VE/V-MCM-41/CF composites also showed high thermal stability. Based on the experimental results, the T10 of the composites achieved a 19.83 °C increment and the char yield was increased from 30.72% to 55.92%. Therefore, this multifunctional material is expected to play an important role in enhancing interfacial strength of composites due to the unusual characteristics of mesoporous silicas; this material would be used in multifunctional carbon fiber composites.
Acknowledgements
The authors acknowledge The Jilin Province Science and Technology Innovation and Achievements Transformation Project of China (20140306011 GX) for financial support.
References
- K. K. C. Ho, G. Beamson, G. Shia, N. V. Polyakova and A. Bismarck, J. Fluorine Chem., 2007, 128, 1359–1368 CrossRef CAS.
- C. Soutis, Prog. Aeronaut. Sci., 2005, 41, 143–151 CrossRef.
- K. Shiba, M. Tagaya, S. Samitsu and S. Motozuka, ChemPlusChem, 2014, 79, 197–210 CrossRef CAS.
- W. H. Liao, H. W. Tien, S. T. Hsiao, S. M. Li, Y. S. Wang, Y. L. Huang, S. Y. Yang, C. C. Ma and Y. F. Wu, ACS Appl. Mater. Interfaces, 2013, 5, 3975–3982 CAS.
- Z. Jiang, H. Zhang, Z. Zhang, H. Murayama and K. Okamoto, Composites, Part A, 2008, 39, 1762–1767 CrossRef.
- S.-L. Gao, E. Mäder and S. F. Zhandarov, Carbon, 2004, 42, 515–529 CrossRef CAS.
- C. Wang, Y. Li, L. Tong, Q. Song, K. Li, J. Li, Q. Peng, X. He, R. Wang, W. Jiao and S. Du, Carbon, 2014, 69, 239–246 CrossRef CAS.
- S. Tiwari, J. Bijwe and S. Panier, Wear, 2011, 271, 2252–2260 CrossRef CAS.
- K. Ma, B. Wang, P. Chen and X. Zhou, Appl. Surf. Sci., 2011, 257, 3824–3830 CrossRef CAS.
- U. Galan, Y. Lin, G. J. Ehlert and H. A. Sodano, Compos. Sci. Technol., 2011, 71, 946–954 CrossRef CAS.
- Y. Yang, C. Lu, X. Su and X. Wang, J. Mater. Sci., 2007, 42, 6347–6352 CrossRef CAS.
- Y. Li, Q. Peng, X. He, P. Hu, C. Wang, Y. Shang, R. Wang, W. Jiao and H. Lv, J. Mater. Chem., 2012, 22, 18748 RSC.
- X. He, C. Wang, L. Tong, R. Wang, A. Cao, Q. Peng, S. Moody and Y. Li, Carbon, 2012, 50, 3782–3788 CrossRef CAS.
- G. Lee, K. D. Ko, Y. C. Yu, J. Lee, W.-R. Yu and J. H. Youk, Composites, Part A, 2015, 69, 132–138 CrossRef CAS.
- M. Sharma, S. Gao, E. Mäder, H. Sharma, L. Y. Wei and J. Bijwe, Compos. Sci. Technol., 2014, 102, 35–50 CrossRef CAS.
- S. Wang, M. Zhang and W. Zhang, ACS Catal., 2011, 1, 207–211 CrossRef CAS.
- A. M. El-Toni, M. A. Habila, M. A. Ibrahim, J. P. Labis and Z. A. Alothman, Chem. Eng. J., 2014, 251, 441–451 CrossRef CAS.
- C. Argyo, V. Weiss, C. Bräuchle and T. Bein, Chem. Mater., 2014, 26, 435–451 CrossRef CAS.
- Y. Zhu, W. Meng, H. Gao and N. Hanagata, J. Phys. Chem. C, 2011, 115, 13630–13636 CAS.
- M. Seifollahi Bazarjani, M. Hojamberdiev, K. Morita, G. Zhu, G. Cherkashinin, C. Fasel, T. Herrmann, H. Breitzke, A. Gurlo and R. Riedel, J. Am. Chem. Soc., 2013, 135, 4467–4475 CrossRef CAS PubMed.
- X. Li, L. Yang, Y. Lei, L. Gu and D. Xiao, ACS Appl. Mater. Interfaces, 2014, 6, 19978–19989 CAS.
- H. Hu, H. Cheng, Z. Liu, G. Li, Q. Zhu and Y. Yu, Nano Lett., 2015, 15, 5116–5123 CrossRef CAS PubMed.
- X. Fang, L. Qiao, G. Yan, P. Yang and B. Liu, Anal. Chem., 2015, 87, 9360–9367 CrossRef CAS PubMed.
- N. Wang, K. Cheng, H. Wu, C. Wang, Q. Wang and F. Wang, Prog. Org. Coat., 2012, 75, 386–391 CrossRef CAS.
- M. Run, S. Wu, D. Zhang and G. Wu, Polymer, 2005, 46, 5308–5316 CrossRef CAS.
- P. Agnihotri, S. Basu and K. K. Kar, Carbon, 2011, 49, 3098–3106 CrossRef CAS.
- H. Wu, B. Tang and P. Wu, J. Phys. Chem. C, 2012, 116, 2246–2252 CAS.
- T. Wang, W. Ma, J. Shangguan, W. Jiang and Q. Zhong, J. Solid State Chem., 2014, 215, 67–73 CrossRef CAS.
- K. Zhang, L. L. Xu, J. G. Jiang, N. Calin, K. F. Lam, S. J. Zhang, H. H. Wu, G. D. Wu, B. Albela, L. Bonneviot and P. Wu, J. Am. Chem. Soc., 2013, 135, 2427–2430 CrossRef CAS PubMed.
- W. Song, A. Gu, G. Liang and L. Yuan, Appl. Surf. Sci., 2011, 257, 4069–4074 CrossRef CAS.
- G. Zhang, S. Sun, D. Yang, J.-P. Dodelet and E. Sacher, Carbon, 2008, 46, 196–205 CrossRef CAS.
- S. Ozcan, J. Tezcan, B. Gurung and P. Filip, J. Mater. Sci., 2011, 46, 38–46 CrossRef CAS.
- H. Yuan, C. Wang, S. Zhang and X. Lin, Appl. Surf. Sci., 2012, 259, 288–293 CrossRef CAS.
- S.-Y. Yang, C.-C. M. Ma, C.-C. Teng, Y.-W. Huang, S.-H. Liao, Y.-L. Huang, H.-W. Tien, T.-M. Lee and K.-C. Chiou, Carbon, 2010, 48, 592–603 CrossRef CAS.
- T. I. T. Okpalugo, P. Papakonstantinou, H. Murphy, J. McLaughlin and N. M. D. Brown, Carbon, 2005, 43, 153–161 CrossRef CAS.
- M. S. Islam, Y. Deng, L. Tong, S. N. Faisal, A. K. Roy, A. I. Minett and V. G. Gomes, Carbon, 2016, 96, 701–710 CrossRef CAS.
- C. Wang, X. Ji, A. Roy, V. V. Silberschmidt and Z. Chen, Mater. Des., 2015, 85, 800–807 CrossRef CAS.
- T. Chen, B. Du and Z. Fan, Langmuir, 2012, 28, 15024–15032 CrossRef CAS PubMed.
- L. Liu, L. Xiao, X. Zhang, M. Li, Y. Chang, L. Shang and Y. Ao, RSC Adv., 2015, 5, 57853–57859 RSC.
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 ,
Lei Shang,
Linghan Xiao* and
Yuhui Ao*
,
Lei Shang,
Linghan Xiao* and
Yuhui Ao*
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :
: