
Facile fabrication of stable and high-rate Si/NiSix/CNTs Li-ion anodes with a buffering interface†
Xu Fanab,
Jingjing Jia,
Xiangping Jiangb,
Wei Wang*a and
Zhaoping Liu*a
aNingbo Institute of Materials Technology and Engineering, Chinese Academy of Sciences, Ningbo, Zhejiang 315201, P. R. China. E-mail: wangwei@nimte.ac.cn; liuzp@nimte.ac.cn
bDepartment of Material Science and Engineering, Jiangxi Key Laboratory of Advanced Ceramic Materials, Jingdezhen Ceramic Institute, Jingdezhen, 333001, Jiangxi, China
First published on 15th August 2016
Abstract
Silicon (Si)/carbon nanotubes (CNTs) composites are ideal anode materials for lithium ion batteries. However, due to the large volume expansion mismatch between Si and CNTs, the cycle life of conventional Si/CNTs composites fabricated by different approaches is still very limited. Here, a novel Si/nickel-Si alloy (NiSix)/CNTs composite has been successfully designed and fabricated through a chemical vapor deposition method with the assistance of uniformly embedded Ni nanoparticles. The CNTs grow up from the composite and serve as strong rivets, which greatly improves the electrical connection stability at the interface between Si and CNTs. Consequently, the corresponding discharge capacity can remain at around 550 mA h g−1 with a capacity retention of 84% for 300 cycles, while a high rate capability can be achieved with a discharge capacity of 420 mA h g−1 at 4 A g−1.
1. Introduction
Silicon (Si) is one of the most attractive anode materials for lithium ion (Li-ion) batteries due to its well-known high theoretical capacity of about 4200 mA h g−1, which is ten times higher than that of commercial graphite anodes.1,2 However, Si anodes suffer from a large volume change with a percentage of 300%, which prevents them from being used in practical applications.3 The volume change causes the pulverization of the active material, and the electrical disconnection from the current collector, which lead to rapid capacity fade.4,5 It was widely accepted that a Si-based composite mixed with some inactive materials or carbon can be very useful for accommodating the volume expansion. Since the pulverization of Si parts cannot be avoided in this approach, the resulting cycle life of the Si-based electrode is still limited. Detailed research indicates that Si nanoparticles can survive during the repeated expansion/shrinking process, if their size is lower than 150 nm.6 Numerous Si nanostructures have been developed with the purpose of improving the battery performance, such as nanospheres, nanowires, nanotubes, and porous three-dimensional (3D) structures.7–10 The preparation of porous Si is another alternative way to enhance the stability of battery due to it can provide sufficient space to absorb the large volume expansion.11 Various methods, such as chemical/electrochemical etching and porous templates have been employed to prepare porous Si with significant improved battery performance.12–14 However, the practical applications of these Si-based materials were hampered by the complicated, expensive or dangerous preparation process.More recently, there is a trend to combine all the advantages in these approaches to construct porous nanostructured Si composite with carbon. For example, Cui et al. reported an impressive ultra-long cycle life of over 1000 cycles by embedding nano-Si in hollow carbon fibers.15 The core–shell Si/C hollow fibers can also be fabricated by an electro-spun method which created numerous pores between Si nanoparticles to accommodate the volume expansion.16 On the other hand, the Si nanoparticles can also be easily immersed into porous carbon matrix by simply pre-embedding some SiO2 particles as sacrificing units to generate the pores after etching process. As previously reported in our group, the porous Si/C composites could be fabricated in a large scale by spray drying method, which also exhibited good electrochemical performance.17,18 Among various porous Si/C composites, the synthesis of Si/CNTs composites is considered as one of the most promising choices because of CNTs have ultra-high surface area and high electrical conductivity. Various Si/CNTs composites have been successfully fabricated by mixing CNTs and Si particles, coating Si on CNTs through chemical vapor deposition (CVD) and growing CNTs on Si particles etc. High capacity has been demonstrated in these free-standing Si/CNTs paper, Si/CNTs sponge and Si/MWNT nanocomposites.19–21 However, although the Si and CNT can be mixed in nanosize level in above methods, the cycle life of these materials is still limited within 100 cycles. The detailed study indicates that the large volume expansion mismatch at the interface between Si and CNTs leads to the formation of pores at the interface, which might be the main reason causing capacity fade.16 Several approaches have been developed to build the buffering interface, which has significantly improved the cycle life. These include preparing rough surface at the interface, employing buffering materials with in-between expansion, and using flexible conducting substrate.22–28 However, all the methods reported can only be used in the plan system, which are not suitable for the conventional Li-ion battery electrodes.
Herein, a novel Si/NiSix/CNTs composite was designed and fabricated by in situ growing CNTs on the Si nanoparticles with co-precipitated Ni(OH)2 as catalyst. It is found that as-grown CNTs have a much stronger bonding force by embedding the CNTs into the composite with the formation of a buffering interface. Thus, the structural stability would be improved greatly, and the cycle life would be enhanced significantly accordingly.
2. Experimental
Fig. 1 presents the typical fabrication process of Si/NiSix/CNTs composites. 50 g Si powders (Sinopharm Chemical Reagent Co., Ltd, 81012160) were milled with 1000 g deionized (DI) water for 12 hours by a high-speed ball-milling machine. It is worth to mention that the Si nanoparticles can be manufactured in large scale of over one kilogram in one batch by a conventional ball-milling process (Fig. S1†). A certain amount of the as-obtained Si nanoparticle slurry was subsequently mixed with 0.5 g super-P (SP) (Shanghai hui industrial chemical co, LTD) and 50 g ammonia. After stirring for 2 hours, nickel acetate solution (0.5 mol L−1) was dropped into the mixture. Then the mixture was dried in an oven at 80 °C for 48 hours and then loaded in a corundum boat in a tube furnace for CVD process. With the protection of argon gas of 200 mL min−1 and H2 of 15 mL min−1, the sample was heated to the temperature of 900 °C at a rate of 10°C min−1. Then, C2H4 of 200 mL min−1 was introduced into the furnace for 30 minutes. Finally, Si/NiSix/CNTs composites were obtained by HNO3 (3 mol L−1) treatment and subsequent washing with DI water three times. For comparison, Si/NiSix/CNTs composites with different contents of Ni were prepared. The final weight ratio of Si atoms and Ni atoms is 97![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 (named 3 wt% Ni) and 4:1 (named 20 wt% Ni). A conventional Si/CNTs composite by simply mixing Si nanoparticles with CNTs was also fabricated using the same CVD process.
:3 (named 3 wt% Ni) and 4:1 (named 20 wt% Ni). A conventional Si/CNTs composite by simply mixing Si nanoparticles with CNTs was also fabricated using the same CVD process.
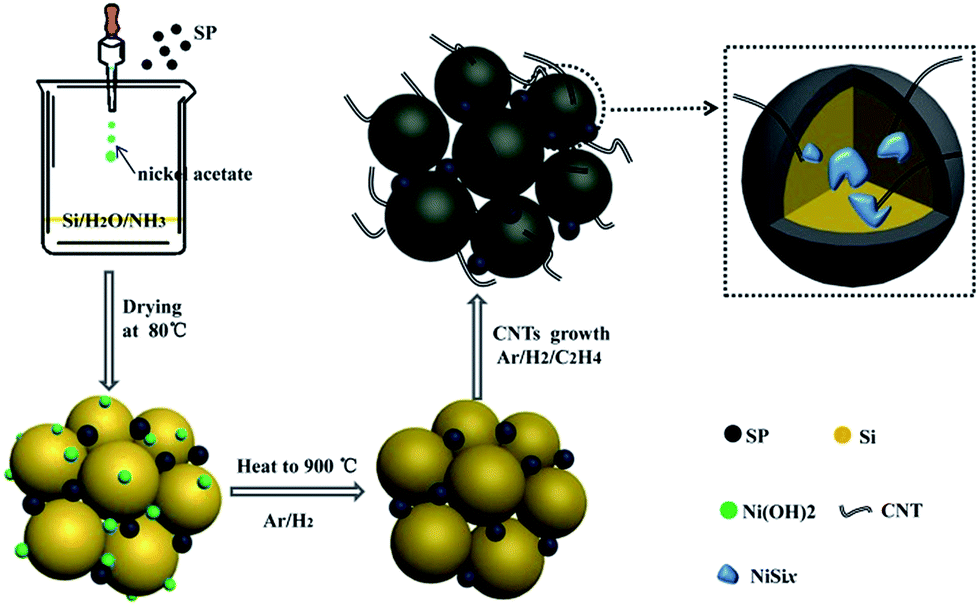 | ||
| Fig. 1 Schematic for preparation procedures of Si/NiSix/CNTs composite. | ||
The surface morphology of all products was investigated by field emission scanning-electron microscopy (FE-SEM, Hitachi S-4800) and transmission electron microscopy (TEM, FEI Tecnai G2 F20). The Brunner–Emmet–Teller (BET) measurements were conducted on a Micromeritics ASAP-2020 M nitrogen adsorption apparatus. Pore size distribution plot was obtained by the Barrett–Joyner–Halenda (BJH) method. Powder X-ray diffraction (XRD) measurements were analyzed by an AXS D8 Advance diffractometer (Cu Kα radiation; receiving slit, 0.2 mm).
The anodes were prepared by mixing the as-prepared composites, super P conductive carbon black and alginate in a weight ratio of 80:10:10 with DI water as the solvent. The resulting slurry was casted onto copper foil, and then dried at 80 °C under vacuum for 12 hours. Electrochemical measurements were carried out in CR2032-type coin cells which were assembled in an argon filled glove box. DMC based electrolyte (Zhang jia gang Guo tai Huarong Chemical New Material Co., Ltd., S-3215G) and separator (Celgard, 2025) were used. All battery cells were activated at a current density of 50 mA g−1 for the first three cycles, and then cycled at different rate for the rest cycles within the voltage window of 0.005–1.5 V by using a battery test system (Jinnuo Wuhan Corp., LANDCT2001A).
3. Results and discussion
As shown in Fig. 2a, the Si/Ni(OH)2 precursor was in form of various microsized particles with a broad size distribution due to the random co-precipitation and the agglomeration in the following drying process. It is hard to distinguish the Si nanoparticles in the close-up image in the inset of Fig. 2a. After the CVD process at 900 °C, the as-obtained composites remain the similar shapes with additional fibers on surface (Fig. 2b). The careful review in the inset image of Fig. 2c clearly presents the growth of amounts of fiber structures. The corresponding TEM characterization (Fig. 2c) demonstrates its nature of carbon nanotubes, which is similar to other reports with Ni catalyst in CVD process. The CNTs grew up from the inside of composites, which have diameters of 20–40 nm and lengths of 1–2 μm. The adherence between CNTs and composite is so strong that they cannot be detached from composites by high power ultrasonication. The XRD results of bare Si, Si/Ni(OH)2 precursor and Si/NiSix/CNTs composites were seen in (Fig. 2d). The sharp peaks located at 28.4, 47.3, 56.1, 58.8, 69.1 and 88.0° can be well indexed to the (111), (220), (311), (400), (331) and (422) planes of crystal silicon (JCPDS no. 27-1402), respectively. Three additional peaks located at 44.48, 51.83, and 76.35° might correspond to the diffraction of NiSix (JCPDS no. 89-5155) or Ni (JCPDS no. 70-1849). As mentioned in the Experimental section, all the samples were washed by HNO3 solution to remove Ni catalyst. During this etching process, a lot of bubbles appear at the beginning process, and gradually disappear at last. Thus, it is believed that all the Ni has been completely removed, and the remained Ni elements should be in the form of NiSix. EDX analysis on the sample after HNO3 treatment indicates that the weight ratio of Si and Ni kept at about 4:1 (Fig. S2†). The carbon content is about 40 wt% in the final composite based on the TGA result (Fig. S3 in ESI†). In addition, the specific surface area is significantly enhanced from 26.2 m2 g−1 to 269 m2 g−1 after the CVD process. As shown in Fig. 2f, the pore size distribution exhibits a hierarchical pore structure with a peak of mesopores centered at around 3 nm, which might originate from the growth of CNTs and the deposition of carbon. The large specific surface area and porous structures can facilitate the penetration of the electrolyte, and provide a great amount of contact area between the electrolyte and the electroactive surface of the materials. More importantly, the increase of pores is good for accommodating the volume changes of Si nanoparticles during the charge/discharge process, and hence more stable architecture and longer cycle life can be expected.
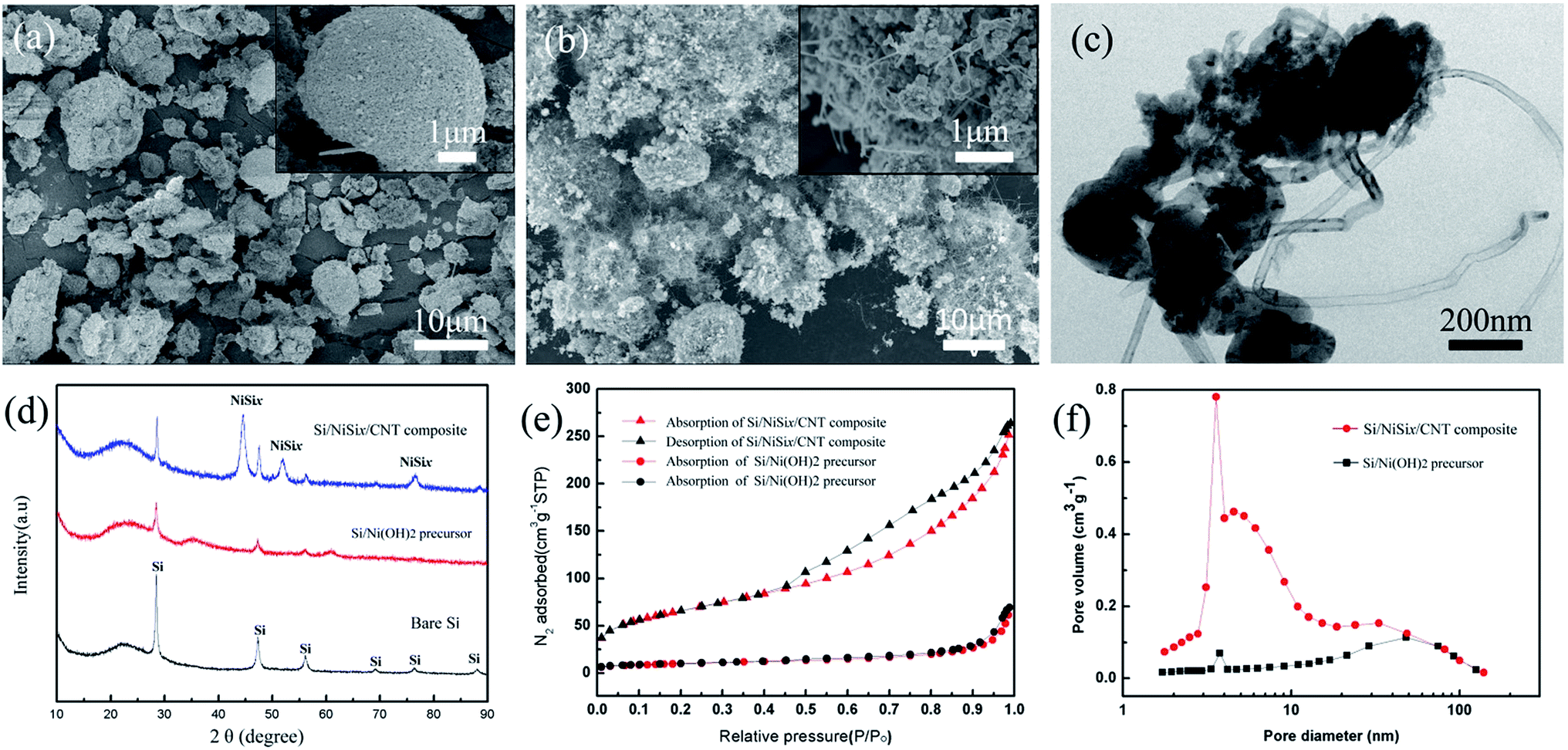 | ||
| Fig. 2 SEM images of (a) Si/Ni(OH)2 precursor and (b) Si/NiSix/CNTs composites, inset images present the close-up morphologies. (c) TEM image of Si/NiSix/CNTs composites. (d) X-ray diffraction patterns of bare Si, Si/Ni(OH)2 precursor and Si/NiSix/CNTs composites. (e) Nitrogen adsorption–desorption isotherm linear plots and (f) pore size distribution of Si/Ni(OH)2 precursor and Si/NiSix/CNTs composite. | ||
The electrochemical properties of all the prepared anodes were studied in the range of 0.005–1.5 V (versus Li+/Li) at the current density of 0.4 A g−1, except the initial three cycles with 0.05 A g−1 for activation. A simple mixture of Si and CNTs usually leads to a high discharge capacity of over 1000 mA h g−1 at a current density of 0.05 Ag−1 (Fig. 3a). However, the discharge capacity can be remained at only 200 mA h g−1 after 50 cycles. With introducing 3 wt% Ni as the catalyst, the resulted Si/NiSix/CNTs composite exhibits a high discharge capacity of above 1200 mA h g−1 for the first three cycles at a current density of 0.05 A g−1. Due to the increased current density of 0.4 A g−1 starting from the forth cycle, a lower discharge capacity of around 950 mA h g−1 appears, and gradually decreases to 650 mA h g−1 after 100 cycles. The capacity retention is significantly better than that of the mixture of Si and CNTs. However, it is still far away from the practical applications with an expected cycle life of hundreds of cycles with capacity retention of 80%. After increasing the Ni content to 20 wt%, the corresponding discharge capacity is around 800 mA h g−1 at the current density of 0.05 A g−1, which is a little lower than that of Si/NiSix/CNTs composite with 3 wt% Ni due to the increase of inactive Ni content. As a benefit, the discharge capacity can be much more stable at around 650 mA h g−1 after increasing the current density to 0.4 A g−1. After 300 charge/discharge cycles, the discharge capacity can be remained at 550 mA h g−1. In other words, the cycle life reaches longer than 300 cycles with the capacity retention of 80%, which represents great potential towards the practical applications.
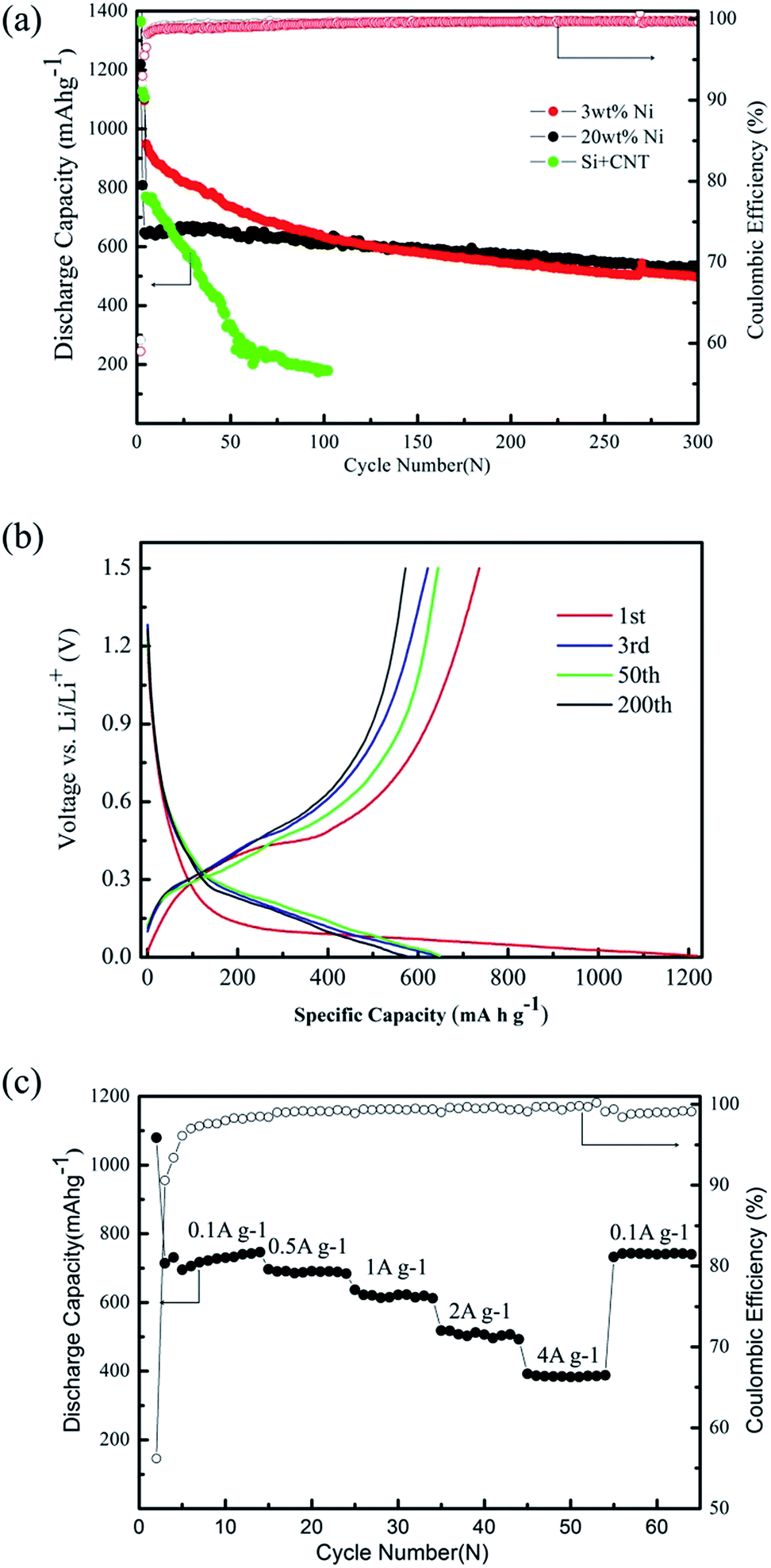 | ||
| Fig. 3 (a) Cycling performance of different Ni contention at a current density of 0.05 A g−1 in the first three cycles and at 0.4 A g−1 in the following cycles. (b) Galvanostatic charge/discharge profiles of the Si/NiSix/CNTs composite for the 1st, 3rd, 50th and 200th cycle. (c) Rate performance of Si/NiSix/CNTs composite. | ||
The corresponding voltage profiles of the Si/NiSix/CNTs composite anode with 20% Ni for the 1st, 3rd, 50th and 200th galvanostatic charge/discharge cycles are shown in Fig. 3b. In the first cycle, the discharge and charge capacities are 1218 and 735.9 mA h g−1 respectively with a coulombic efficiency of 60.4%. The low efficiency could be mainly due to the larger surface area of the Si/NiSix/CNTs network, and formation of an irreversible solid electrolyte interphase (SEI) layer in the first discharge process, which might be improved by surface modification or prelithiation etc.29,30 A long flat plateau below 0.1 V appears in the first discharge process, which contributes to the Li-alloying process of crystalline Si to amorphous LixSi.9 During the next cycles, the discharge and charge curves show the stable characteristic of amorphous Si with a reversible capacity around 600 mA h g−1 at high coulombic efficiency of more than 99%.
Besides the significant enhancement of cycle life, the resulted Si/NiSix/CNTs composite also exhibit a surprising rate capability. The electrode was cycled at current density of 0.1 A g−1, 0.5 A g−1, 1 A g−1, 2 A g−1 and 4 A g−1 every ten cycles (Fig. 3c), except the first three cycles at low current density of 0.05 A g−1. The discharge capacity slightly decreases from 680 mA h g−1 to 420 mA h g−1 as the current density is increased from 0.1 A g−1 to 4 A g−1. It means that a full discharge process can be completed within 6 minutes while the capacity is still higher than the theoretical charge capacity of commercial graphite (372 mA h g−1). The discharge capacity can be recovered if the current rate is changed to 0.1 A g−1 subsequently, which indicates the excellent cycling stability and rate capability of the Si/NiSix/CNTs composite.
Actually, the in situ growth of CNTs for improving the battery performance of Si based anodes has been investigated in other reports. The Fe-catalyzed growth of CNTs just on Si nanoparticles surface can present a high capacity of 1500 mA h g−1, but a quite short cycle life of about 100 cycles.21 Compared to that, the significantly improved cycle life and high-rate performance in this work attract intensive attention for a deep understanding, which might raise the new light for further improving the battery performance of Si anodes in a simple, but versatile way. The element distribution of Si, Ni, C (Fig. 4a–d) in a typical Si/NiSix/CNTs composite, which indicates that Ni was embedded inside of the particles. It is worth to mention that some dark nanoparticles can be easily observed at the root of CNTs due to the obvious contrast difference (more images are presented in the ESI, Fig. S4†). Compared to the growth of CNTs on surface of Si nanoparticles, herein NiSix nanoparticles are embedded with CNTs penetrating inside the composite as shown in the EDS mapping (Fig. 4e–h), CNTs with NiSix nanoparticles inside the composite might have a stronger connection like the rivet structure, and meanwhile the NiSix at the interface can serve as the buffering interface. With careful observation by HRTEM (Fig. S5†), it shows that a thin layer carbon was also deposited on surface for a better covering. It is well-known that there is a large volume expansion mismatch at the interface between carbon (9%) and Si (300%). Especially, the root at the interface of 1D structure and substrate is always the most weaken part.31,32 Thus, the simple connection by in situ growth of CNTs on surface of Si particles would not be able to survive after repeated electrochemical cycling (Fig. 4i). Although many methods have been developed to buffer the expansion mismatch at the interface, few approaches can work on the conventional powder composites. In this work, the as-grown CNTs with Ni as catalyst are of big diameter throughout the composite which can serve as the strong rivets to activate the electrical connection between particles. In addition, the carbon coating on surface can further ensure the binding strength between CNTs and Si particles for a better toleration on the expansion mismatch at the interface. In Conclusion, this unique architecture with Ni catalyst, CNTs as electrical rivets, and carbon coating as binding layer significantly improves the structural stability at the interface, and hence, the remarkably prolonged electrochemical cycle life.
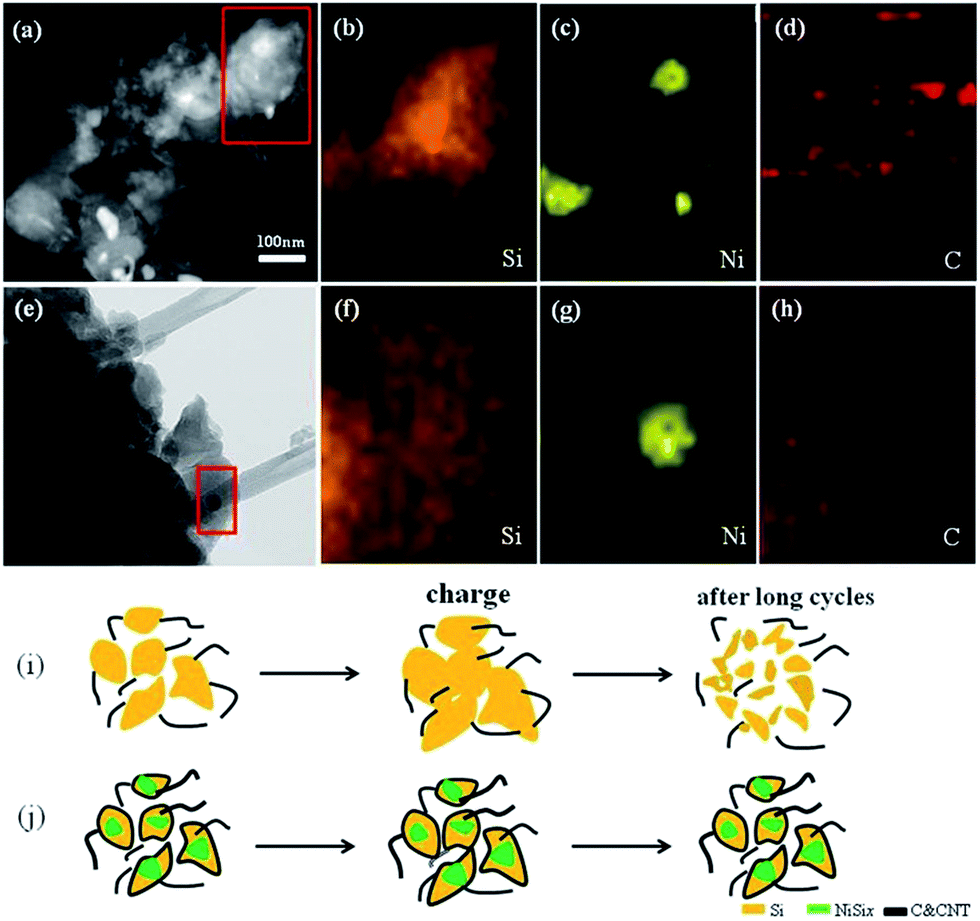 | ||
| Fig. 4 TEM images (a and e) of Si/NiSix/CNTs composite. (b–d) EDX mapping data of Si, Ni, C, respectively corresponded of red box in (a). (f–h) EDX mapping data of Si, Ni, C, respectively corresponded of red box of (e). Schematic of possible structure evolution during electrochemical process for (i) in situ growth of CNTs on surface of Si particles and (j) Si/NiSix/CNTs composite with buffering interface. | ||
4. Conclusions
A novel Si/NiSix/CNTs composite is successfully designed and fabricated through a facile in situ CNTs growth approach. The CNTs growing up from the composite and a thin layer carbon coating on surface could greatly improve the electrical connection by buffering the expansion mismatch at the interface between CNTs and Si particles. Discharge capacity can be remained at about 550 mA h g−1 for 300 cycles with capacity retention of 84%. Meanwhile, high-rate performance was also achieved with discharge capacity of 420 mA h g−1 at the current density of 4 A g−1.Acknowledgements
This work is supported by the National Natural Science Foundation of China (51402322), and the Natural Science Foundation of Zhejiang (LY13A040005).References
- B. A. Boukamp, G. C. Lesh and R. A. Huggins, J. Electrochem. Soc., 1981, 128, 725–729 CrossRef CAS.
- J. R. Dahn, T. Zheng, Y. H. Liu and J. S. Xue, Science, 1995, 270, 590–593 CAS.
- J. O. Besenhard, J. Yang and M. Winter, J. Power Sources, 1997, 68, 87–90 CrossRef CAS.
- W. Weng, H. J. Lin, X. L. Chen, J. Ren, Z. T. Zhang, L. B. Qiu, G. Z. Guan and H. S. Peng, J. Mater. Chem. A, 2014, 2, 9306–9312 CAS.
- I. Kovalenko, B. Zdyrko, A. Magasinski, B. Hertzberg, Z. Milicev, R. Burtovyy, I. Luzinov and G. Yushin, Science, 2011, 334, 75–79 CrossRef CAS PubMed.
- X. H. Liu, L. Zhong, S. Huang, S. X. Mao, T. Zhu and J. Y. Huang, ACS Nano, 2012, 6, 1522–1531 CrossRef CAS PubMed.
- Y. Yao, M. T. McDowell, I. Ryu, H. Wu, N. A. Liu, L. B. Hu, W. D. Nix and Y. Cui, Nano Lett., 2011, 11, 2949–2954 CrossRef CAS PubMed.
- H. Ma, F. Y. Cheng, J. Chen, J. Z. Zhao, C. S. Li, Z. L. Tao and J. Liang, Adv. Mater., 2007, 19, 4067 CrossRef CAS.
- L. F. Cui, R. Ruffo, C. K. Chan, H. L. Peng and Y. Cui, Nano Lett., 2009, 9, 491–495 CrossRef CAS PubMed.
- M. H. Park, M. G. Kim, J. Joo, K. Kim, J. Kim, S. Ahn, Y. Cui and J. Cho, Nano Lett., 2009, 9, 3844–3847 CrossRef CAS PubMed.
- J. Cho, J. Mater. Chem., 2010, 20, 4009–4014 RSC.
- H. J. Tian, X. J. Tan, F. X. Xin, C. S. Wang and W. Q. Han, Nano Energy, 2015, 11, 490–499 CrossRef CAS.
- H. C. Tao, L. Z. Fan, W. L. Song, M. Wu, X. B. He and X. H. Qu, Nanoscale, 2014, 6, 3138–3142 RSC.
- X. K. Huang, J. Yang, S. Mao, J. B. Chang, P. B. Hallac, C. R. Fell, B. Metz, J. W. Jiang, P. T. Hurley and J. H. Chen, Adv. Mater., 2014, 26, 4326–4332 CrossRef CAS PubMed.
- H. Wu, G. Y. Zheng, N. A. Liu, T. J. Carney, Y. Yang and Y. Cui, Nano Lett., 2012, 12, 904–909 CrossRef CAS PubMed.
- T. H. Hwang, Y. M. Lee, B. S. Kong, J. S. Seo and J. W. Choi, Nano Lett., 2012, 12, 802–807 CrossRef CAS PubMed.
- D. S. Jung, T. H. Hwang, S. B. Park and J. W. Choi, Nano Lett., 2013, 13, 2092–2097 CrossRef CAS PubMed.
- Z. Z. Li, W. Wang, Z. H. Li, Z. H. Qin, J. Wang and Z. P. Liu, J. Power Sources, 2015, 286, 534–539 CrossRef CAS.
- Q. Z. Xiao, Y. Fan, X. H. Wang, R. A. Susantyoko and Q. Zhang, Energy Environ. Sci., 2014, 7, 655–661 CAS.
- L. B. Hu, H. Wu, Y. F. Gao, A. Y. Cao, H. B. Li, J. McDough, X. Xie, M. Zhou and Y. Cui, Adv. Energy Mater., 2011, 1, 523–527 CrossRef CAS.
- P. F. Gao, Y. Nuli, Y. S. He, J. Z. Wang, A. I. Minett, J. Yang and J. Chen, Chem. Commun., 2010, 46, 9149–9151 RSC.
- S. C. Zhang, Z. J. Du, R. X. Lin, T. Jiang, G. R. Liu, X. M. Wu and D. S. Weng, Adv. Mater., 2010, 22, 5378 CrossRef CAS PubMed.
- H. Park, S. Choi, S. Lee, G. Hwang, N. S. Choi and S. Park, J. Mater. Chem. A, 2015, 3, 1325–1332 CAS.
- J. Niu, S. Zhang, Y. Niu, H. H. Song, X. H. Chen, J. S. Zhou and B. Cao, J. Mater. Chem. A, 2015, 3, 19892–19900 CAS.
- R. Krishnan, T. M. Lu and N. Koratkar, Nano Lett., 2011, 11, 377–384 CrossRef CAS PubMed.
- L. W. Ji, H. H. Zheng, A. Ismach, Z. K. Tan, S. D. Xun, E. Lin, V. Battaglia, V. Srinivasan and Y. G. Zhang, Nano Energy, 2012, 1, 164–171 CrossRef CAS.
- X. H. Wang, L. N. Sun, R. A. Susantyoko, Y. Fan and Q. Zhang, Nano Energy, 2014, 8, 71–77 CrossRef CAS.
- S. B. Son, S. C. Kim, C. S. Kang, T. A. Yersak, Y. C. Kim, C. G. Lee, S. H. Moon, J. S. Cho, J. T. Moon, K. H. Oh and S. H. Lee, Adv. Energy Mater., 2012, 2, 1226–1231 CrossRef CAS.
- M. W. Forney, M. J. Ganter, J. W. Staub, R. D. Ridgley and B. J. Landi, Nano Lett., 2013, 13, 4158–4163 CrossRef CAS PubMed.
- L. Q. Mai, L. Xu, B. Hu and Y. H. Gu, J. Mater. Res., 2010, 25, 1413–1420 CrossRef CAS.
- M. Tian, W. Wang, Y. J. Wei and R. G. Yang, J. Power Sources, 2012, 211, 46–51 CrossRef CAS.
- H. W. Liao, K. Karki, Y. Zhang, J. Cumings and Y. H. Wang, Adv. Mater., 2011, 23, 4318–4322 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: SEM images and structure characterization. See DOI: 10.1039/c6ra13620f |
| This journal is © The Royal Society of Chemistry 2016 |