
The effective removal of Cr(vi) ions by carbon dot–silica hybrids driven by visible light †
Abstract
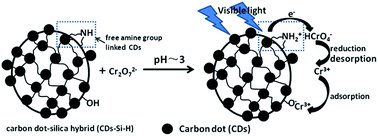
Methods to economically and efficiently remove highly toxic hexavalent chromium (Cr(VI)) from discharged industrial effluents have attracted critical attention due to the carcinogenic, mutagenic and teratogenic effects of Cr(VI). In the present work, we have explored the potential of carbon dots (CDs) in the elimination of Cr(VI) contamination. A series of carbon dot–silica hybrid photocatalysts (CD–Si–H) were firstly prepared by the co-hydrolysis and condensation of amine silane functionalized carbon dots and tetraethyl orthosilicate (TEOS). Due to the action of the carbon dots in the silica matrix, CD–Si–H demonstrated significant visible light absorption and good blue fluorescence with a longer fluorescent lifetime. With the increase of CDs, however, CD–Si–H suffered from a gradual reduction in the mesoporous structure, surface area and pore volume. In spite of this, the incorporation of a high concentration of CDs in the hybrid led to better visible light-harvesting, a stronger ability to capture Cr(VI) ions and a more effective visible-light-driven photo-reduction of Cr(VI). The presence of Cr(VI) resulted in a decrease of the fluorescent lifetime of the CDs embedded in CD–Si–H, suggesting that the electron transfer from the excited CDs to Cr(VI) is responsible for the effective photoreduction of Cr(VI). This work provides new methods to easily synthesize carbon dot–silica hybrids and to eliminate Cr(VI) contaminants under visible light.
 Please wait while we load your content...
Please wait while we load your content...

