
Fabrication of TiO2 nanosheets via Ti3+ doping and Ag3PO4 QD sensitization for highly efficient visible-light photocatalysis
Abstract
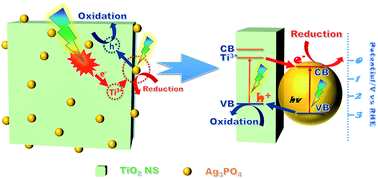
Visible-light photocatalysis has attracted much attention in environmental remediation and sustainable energy utilization, however, it is still a great challenge to develop highly efficient and stable visible-light photocatalyst. Herein, we developed Ag3PO4 quantum dots (QDs) sensitized and Ti3+-doped TiO2 nanosheets (NS) via a solvothermal/in situ precipitation method. The TiO2/Ag3PO4 ratio in the composite was tuned from 3 : 1 to 1 : 4 to optimize the dispersion and size of Ag3PO4 QDs, and the best dispersed Ag3PO4 QDs with the smallest size (ca. 2 nm) was obtained for TA1 : 3. The characterizations confirm that abundant Ti3+ defects are introduced into TiO2, and the interaction between Ag3PO4 QDs and TiO2 NS is in the form of Ag–O–Ti bonds, which benefit the visible-light absorption and accelerates the charge separation. Moreover, the well-matched band structures drive the electrons to Ag3PO4 and holes to TiO2 {001} faces, respectively. Therefore, TA1 : 3 shows a 1.7-fold, 1.4-fold and 5-fold higher activity than bulk Ag3PO4 in MO, phenol photodegradation, and PEC water splitting, respectively. In addition, the sample shows relatively high photostability. Thus, we believe that the rational design of heterostructures based on the matched band and abundant defects can fabricate the highly reactive photocatalysts.
 Please wait while we load your content...
Please wait while we load your content...

