
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffects of protein species and surface physicochemical features on the deposition of nanoparticles onto protein-coated planar surfaces†
Kaoru
Ikuma
ab,
Zhiwei
Shi
c,
Amy V.
Walker
c and
Boris L. T.
Lau
*ad
aDepartment of Geology, Baylor University, Waco, TX 76798, USA
bDepartment of Civil, Construction and Environmental Engineering, Iowa State University, Ames, IA 50011, USA
cDepartment of Materials Science & Engineering, University of Texas at Dallas, Richardson, TX 75080, USA
dDepartment of Civil & Environmental Engineering, University of Massachusetts Amherst, Marston Hall, 130 Natural Resources Road, Amherst, MA 01003-9293, USA. E-mail: borislau@engin.umass.edu; Fax: +1-413-545-2840; Tel: +1-413-545-5423
First published on 3rd August 2016
Abstract
Proteins are often an important component of many bulk surfaces in biological and environmental systems that are coated with complex organic compounds that may also interact with NPs. We investigated the deposition of bare hematite NPs onto various proteins adsorbed on either negatively- or positively-charged bottom surfaces. Bovine serum albumin (BSA), lysozyme, and ubiquitin were used as model proteins and total protein extracts from two bacterial strains, Escherichia coli and Pseudomonas fluorescens, were used as complex protein mixtures. The NP deposition extents and rates were shown to be significantly different depending on the protein. The maximum difference observed was 8.6 ± 3.2 fold between E. coli and P. fluorescens proteins adsorbed onto positively-charged planar surfaces. These differences in NP deposition characteristics are attributed to the differences in physicochemical features of the topmost surface of the protein layer, such as the amino acid profiles, surface charge, and hydrophilicity. Such differences were likely driven by differences in species, orientation, and conformation of the adsorbed proteins. In particular, NP deposition was driven by various combinations of electrostatic and hydrophobic interactions. This study indicates that NP deposition onto surface-adsorbed proteins is an important mechanism in protein–NP interactions and that the deposition is strongly dependent on both the conformation and chemical characteristics of the adsorbed protein layer.
Introduction
The interactions between proteins and nanoparticles (NPs) in aqueous environments have been extensively studied in recent years with a focus on the “protein corona” that forms when NPs are exposed to physiological fluids.1 Many variables such as the type and surface characteristics of NPs and proteins as well as solution conditions have been shown to affect the resulting corona (as reviewed by several researchers2–6). While such interactions in bulk suspension are important for assessing NP exposure to biologically-relevant systems, another likely but understudied mode of contact between NPs and proteins is the interfacial interaction at a planar surface. Based on their small size, NPs are come into contact with complex surfaces that appear planar from the NP point of view. Since proteins are known to readily adsorb onto solid surfaces in many environments within biological, engineered, and natural systems (see for example ref. 7–10), the interfacial interactions between NPs and bulk surfaces are significantly impacted by the presence of protein layers. Such interfacial interactions are important not only in biological systems in which proteins are abundant, but also in environmental systems that contain proteins as major parts of natural organic matter7 or biofilms.8,9 In these systems, the interfacial deposition of NPs onto surface-adsorbed proteins can greatly influence the transport of NPs and their ultimate fate.Surface adsorption of proteins can result in various protein conformations and orientations, depending on the surface and water chemistry conditions. These differences in protein immobilization will likely result in significantly different characteristics for the topmost surface of the protein layer, which is the surface that is directly accessible for deposition, and can change the likelihood of subsequent NP deposition. The complex interplay of protein immobilization, surface physicochemical characteristics, and NP characteristics involved in such protein–NP interactions is a challenging yet important area of study. Research directly investigating the interactions between NPs and surface-adsorbed proteins is currently lacking; however, previous studies have reported changes in the adsorption of micron-sized particles10,11 and bacterial cells12 onto surface-adsorbed proteins due to differences in surface chemistry such as charge distribution and hydrophobicity. In particular, the surface coverage of adsorbed proteins were shown to result in surface heterogeneity that significantly impacted microparticle attachment.10 These non-NP studies point to the importance of various physicochemical parameters that are likely to greatly influence NP interactions with surface-bound proteins. Furthermore, the impacts of solution conditions, including ionic strength and pH, have been demonstrated to influence NP deposition onto complex organic-coated surfaces such as natural organic matter13,14 and biofilms;15,16 these effects are relatively well-understood and are expected to impact NP deposition onto surface-bound proteins in specific ways. However, the effects of different characteristics of the organic layer, in particular the orientation and conformation of adsorbed proteins, on NP deposition have not been studied in depth. Therefore, this study was aimed to investigate the interactions between NPs and surface-adsorbed proteins with a specific focus on the impact of different surface characteristics of the protein layer.
In this study, we examined the deposition of positively-charged bare hematite (αFe2O3) NPs onto bulk planar surfaces coated with various proteins by quartz crystal microgravimetry (QCM). While in many relevant environments, NPs are also likely be coated with proteins or organic matter, we used bare NPs in this study as a first step to specifically investigate the effects of surface-adsorbed proteins on NP deposition. To test the impact of different protein conformations and orientations in the interfacial interactions, proteins were first adsorbed on either negatively- or positively-charged planar surfaces, and NPs subsequently deposited on the protein layers. Three model proteins (bovine serum albumin (BSA), lysozyme, and ubiquitin) and total protein extracts from two bacterial strains (Escherichia coli and Pseudomonas fluorescens) were used to further test the effects of surface physicochemical features of various protein layers on the NP deposition characteristics. The model proteins were chosen based on their differences in isoelectric points (pI of BSA, lysozyme and ubiquitin are 4.9, 9.3, and 6.7, respectively17,18). E. coli was chosen due to its well-studied nature as a model organism and P. fluorescens was used as an environmental bacterium that is prevalent in many natural and engineered systems. Results from NP deposition experiments by QCM were correlated to various protein layer surface characteristics including amino acid chemistry assessed by time-of-flight secondary ion mass spectrometry (TOF SIMS), surface zeta potential measured as streaming potential, surface potential heterogeneity and topography determined using Kelvin probe force microscopy (KPFM), and surface hydrophilicity determined by contact angle measurements.
Experimental
Materials
Hematite (αFe2O3) NPs (pseudo-hexagonal platelets) were prepared as described by Schwertmann and Cornell (2000)19 and were a generous gift from Andrew Madden (University of Oklahoma, Norman, OK). Bovine serum albumin (BSA), lysozyme, and ubiquitin were obtained as purified chemicals from Sigma Aldrich (St. Louis, MO) and used as model proteins. Escherichia coli and Pseudomonas fluorescens cultures were grown to late exponential growth phase in Luria-Bertani broth at room temperature (22–23 °C) with shaking at 150 rpm. Following harvesting and washing, the bacterial total proteins were extracted and purified using ammonium sulfate and trichloroacetic acid (TCA) as described previously.20 In brief, the bacterial cells were lysed by sonication in 10 mM phosphate buffer, followed by ammonium sulfate precipitation (at 25% saturation) of total proteins from the crude extract. The resulting protein suspensions were further treated with 12.4% v/v TCA and washed with acetone for additional purification and to remove ammonium sulfate from the protein sample. All chemicals used in this study were ACS grade and were obtained from Thermo Fisher Scientific (Waltham, MA) or VWR International (Radnor, PA) unless specified otherwise.NP characterization
The hydrodynamic diameters by dynamic light scattering (DLS) and zeta potentials of hematite NPs were determined using a Malvern Zetasizer NS (Worcestershire, UK). The zeta potentials of NPs were calculated from measured electrophoretic mobility values using the Smoluchowski approximation. The average hydrodynamic diameter, TEM-based average diameter, and zeta potential in the test solution, 10 mM NaCl (pH 5.7), were 75.7 ± 0.5 nm, 37 nm, and 25.7 ± 0.6 mV (electrophoretic mobility of 2.02 ± 0.04 μm cm V−1 s−1), respectively. The point of zero charge of the hematite NPs was empirically determined to be 7.7 and the critical coagulation concentration for the NPs was 40 mM NaCl (data not shown). Additional details on hematite NP characteristics such as particle size distribution are reported in Lau et al. (2013).21QCM
A Q-Sense E1 quartz crystal microbalance with dissipation monitoring was used to characterize NP deposition following previously described procedures22 with modifications. Silica quartz crystal sensors (14 mm diameter) with a fundamental resonant frequency of 5 MHz (QSX 303, Q-Sense, Gothenburg, Sweden) were used in this study. In brief, 10 mM sodium 4-(2-hydroxyethyl) piperazine-1-ethanesulfonate (HEPES) (pH 7.4) was used as the initial solution to obtain a stable baseline. The protein layer was subsequently adsorbed with or without precoating with poly-L-lysine (PLL) which adds a positive charge to the sensor surface. The protein concentrations of the solutions used were 100 mg L−1 for BSA and ubiquitin, 50 mg L−1 for lysozyme, and approximately 10 mg L−1 for bacterial total protein extracts. The protein concentrations in solution were observed to not significantly affect the mass of proteins adsorbed on the silica sensors (data not shown). All solutions of organic compounds were made in 10 mM HEPES buffer (pH 7.4). Following a thorough rinsing with clean buffer to remove loosely-bound proteins, 10 mM NaCl (pH 5.7) was introduced until a stable baseline was obtained. Then a 10 mg L−1 working suspension of NPs in 10 mM NaCl was pumped through to interact with the surface-adsorbed proteins. A NaCl solution at pH 5.7 was used for the NP adsorption phase of the experiments because hematite NPs were susceptible to accelerated aggregation in buffers and at other pH values (data not shown). Changes in resonance frequency (Δf) and in resonance dissipation (ΔD) were monitored over time as NPs deposited onto the surface. With the best signal-to-noise ratio, Δf and ΔD obtained from the third overtone are presented in this study. The extent of deposition was determined after the sensor electrode was washed with background electrolyte solution to remove unbound NPs and a new stable frequency reading was reached. The mass deposited on the QCM sensor was calculated using the Sauerbrey equation; the deposited layers were considered to be relatively rigid as observed by the ΔDn/(−Δfn/n) ≪ 4 × 10−7 Hz−1 relationship suggested by Reviakine et al. (2011)23 (representative raw QCM data shown in Fig. S1†). The initial deposition rate was determined by calculating the slope of the linear change in adsorbed mass over the first ten minutes of deposition. The temperature of the solutions in the flow module was maintained at 25 ± 0.02 °C for all experiments. The flow rate was kept constant at 100 μL min−1. All test conditions were run at least in triplicate. Real time DLS measurements were run simultaneously during QCM adsorption experiments to monitor NP size changes over time in each test solution.Protein-coated surface characterization
Statistical analysis
Two-tailed Student's t test was used to compare sample values to control values. Results were reported as significantly different with a p-value < 0.05.Results and discussion
Hematite NP deposition onto protein layers
The total extents and initial rates of positively-charged hematite NP deposition onto protein layers adsorbed on negatively- or positively-charged sensor surfaces were measured by QCM and are shown as values normalized to AFM-based surface areas in Fig. 1, respectively (Fig. S2†). Bare and poly-L-lysine (PLL)-coated silica sensors were used as negatively- and positively-charged surfaces, respectively (surface zeta potentials of −80.5 ± 4.4 and 23.6 ± 7.2 mV, respectively, in 10 mM NaCl at pH 5.7). We note that for the duration of the QCM experiments, the NP sizes were shown to be stable with an average hydrodynamic diameter of 75.7 ± 0.5 nm (Fig. S3†). The total extents of hematite NP deposition onto model protein layers (BSA, lysozyme, and ubiquitin) were significantly influenced by the charge of the bottom sensor surface on which the proteins were adsorbed (Fig. 1a). Specifically, the extent of NP deposition was greater on BSA layers adsorbed on negatively-charged sensor surfaces compared to positively-charged surfaces by 7.8 (±3.2) folds (p < 0.001). Similar trends were observed for NP deposition onto ubiquitin layers, in which ubiquitin adsorbed on a negatively-charged sensor surface resulted in NP deposition that was two orders of magnitude greater (p < 0.001). As lysozyme did not adsorb on a positively-charged sensor surface, hematite NP deposition was observed only onto lysozyme layers adsorbed on negatively-charged surfaces. Additionally, the hematite NP deposition extents were affected by the type of proteins adsorbed on the surface. NPs deposited onto BSA layers to a greater extent than ubiquitin or lysozyme layers on negatively-charged silica sensor surfaces (p < 0.005). Similar to the deposition extent, the initial deposition rates were dependent on the type of surface-adsorbed proteins (Fig. 1b). The initial deposition rates were 1.70 (±0.30) fold greater onto ubiquitin compared to BSA adsorbed on negatively-charged sensor surfaces (p = 0.014). On the other hand, the rates for NPs depositing onto BSA adsorbed onto positively-charged sensor surfaces were 1.42 (±0.25) fold greater than negatively-charged surfaces (p = 0.042). These trends are opposite than observed for the total NP deposition extents (Fig. 1a), suggesting that the total number of deposition sites (which affects the total extent) and the affinity for such sites (which affects the initial rates) are not necessarily correlated. For example, ubiquitin adsorbed on a negatively-charged sensor surface appears to have fewer sites for hematite NP deposition than BSA; however, the deposition sites on the ubiquitin surface may have greater affinity for hematite NPs as observed by the larger initial deposition rate.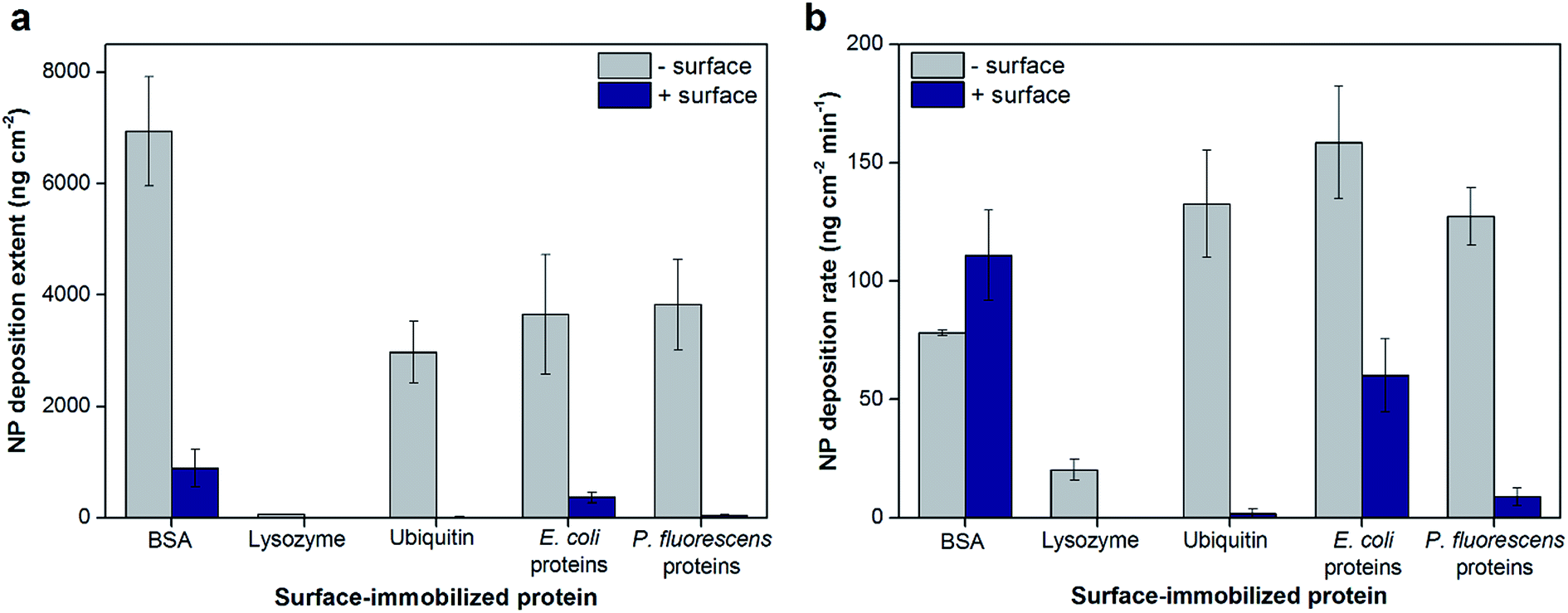 | ||
| Fig. 1 Total hematite NP deposition extents (a) and initial rates (b) onto model and extracted proteins adsorbed on negatively- or positively-charged sensor surfaces in 10 mM NaCl (pH 5.7). Lysozyme only adsorbed onto negatively-charged sensor surfaces; as such, no data were collected for positively-charged surfaces. Values normalized to relative surface areas of the protein layer surfaces determined by AFM for each condition. Error bars indicate the standard deviations of at least triplicate experiments. | ||
The total hematite NP deposition extents and initial rates were similar for protein extracts from E. coli and P. fluorescens adsorbed on negatively-charged sensor surfaces (Fig. 1a and b; p > 0.05) and were comparable to the results for ubiquitin. For protein extracts adsorbed onto positively-charged sensors surfaces, the deposition extents and initial rates were 8.58 (±3.16) and 6.79 (±3.41) fold greater on layers of E. coli proteins compared to P. fluorescens proteins, respectively (p < 0.01). Both the extents and rates of hematite NP deposition were larger by at least 2.6 fold on protein extracts adsorbed on negatively-charged than positively-charged sensor surfaces for both bacterial strains tested (p < 0.01). As the protein extracts are mixtures of many proteins, the identity of the proteins adsorbed on surfaces may be significantly different depending on both the bacterial source and the characteristics of the bottom surface. Even for model proteins, the chemical profile of the topmost surface of the protein layer that is available for NP attachment is expected to be different with varying bottom surfaces. These differences, in turn, change the physicochemical characteristics of the topmost surface of the adsorbed protein layer which has direct effects on the extent and rate of NP deposition. Such characteristics of the protein layers (e.g., surface topography, charge, and surface wettability) and their effects are discussed below.
Effects of the surface physicochemical characteristics of the adsorbed protein layer
The surface roughness of each surface-adsorbed protein layer was analyzed by AFM and reported in Fig. 2. All surfaces appeared to be relatively smooth with the surface roughness ranging only up to 2 nm. For most proteins tested with the exception of BSA, the charge of the bottom sensor surface did not significantly alter the surface roughness of the protein layer (p > 0.05). While the largest difference (2.08 ± 0.80 fold, p = 0.0092) was observed between BSA and ubiquitin layers adsorbed on negatively-charged sensor surfaces, these values varied only by 1 nm. These observations suggest that even though the model proteins vary in their individual sizes, their adsorption onto the QCM sensor surfaces resulted in protein layers with relatively smooth topmost surfaces, suggesting complete coverage of proteins on the sensor surfaces.25 In addition to surface roughness measurements, the QCM-based total mass of each protein layer was comparable to previous studies indicating complete monolayer coverage (e.g., 229 and 679 ng cm−2 for BSA on bare and PLL-coated silica sensors, respectively, were reported in Madliger et al. (2010);27 the BSA masses of adlayers in this study were 368 ± 39 and 655 ± 23 ng cm−2, respectively). As the observed differences in surface roughness were small and no valleys large enough to accommodate the hematite NPs were observed in the AFM images (Fig. S2†), it is likely that the surface topography of the protein layers did not affect hematite NP deposition.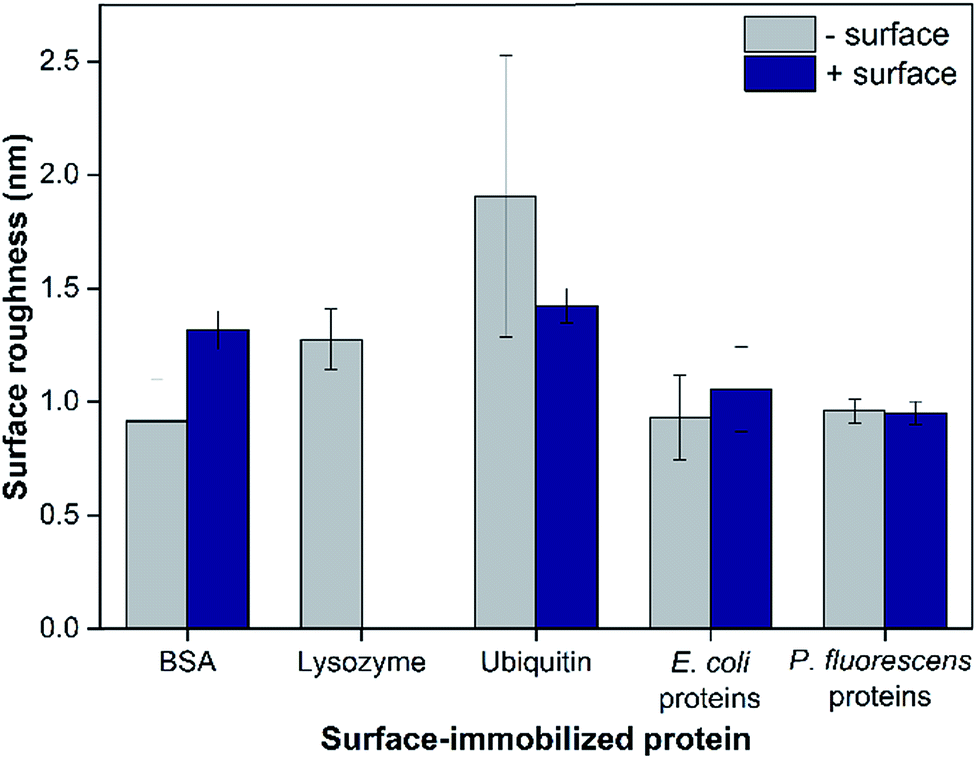 | ||
| Fig. 2 The surface roughness of model and extracted proteins adsorbed on negatively- or positively-charged sensor surfaces based on RMS values analyzed from AFM images. Lysozyme only adsorbed onto negatively-charged sensor surfaces; as such, no data were collected for positively-charged surfaces. Error bars indicate the standard deviations of at least five images. | ||
The surface zeta potentials of surface-adsorbed proteins were calculated based on their streaming potential measurements and are shown in Fig. 2a. All surface zeta potentials were negative even for lysozyme and ubiquitin that have isoelectric points (pI) larger than the tested pH 5.7 (pI of BSA, lysozyme and ubiquitin are 4.9, 9.3, and 6.7, respectively17,18). This discrepancy was expected as pI values are obtained from proteins in bulk solution while the zeta potentials reported herein were based on surface-adsorbed protein layers. Protein adsorption studies have shown that the charge environment of the protein can be greatly distorted upon surface adsorption depending on the conformation and orientation of the immobilized proteins.28 In addition, for all proteins tested, the protein layers adsorbed on negatively-charged sensor surfaces exhibited more negative zeta potentials (1.50 ± 0.12 fold for BSA, 2.04 ± 0.18 fold for ubiquitin, 1.71 ± 0.12 fold for E. coli proteins, and 1.94 ± 0.16 fold for P. fluorescens proteins; p < 0.0001) compared to the same proteins adsorbed onto positively-charged surfaces (p < 0.0001). These observations are likely due to the different conformation and/or orientations of the adsorbed proteins.
Furthermore, the surface zeta potentials of most proteins depended on the bottom surface on which they were adsorbed (p < 0.01). The surface-adsorbed protein extracts from E. coli and P. fluorescens exhibited similar surface zeta potentials on negatively-charged sensors (p = 0.28) but significantly different on positively-charged surfaces (p = 0.00058). These observations may explain the trends seen in the total hematite NP deposition extents onto these protein extract layers (Fig. 1a); the NPs deposited to a greater extent on E. coli protein extracts adsorbed on positively-charged surfaces than P. fluorescens extracts likely due to a stronger charge attraction for positively-charged hematite NPs onto a more negative surface charge. However, the overall correlation between NP deposition extents and surface zeta potentials (Fig. S4†) had no observable trend. On the other hand, the NP deposition rates and surface zeta potentials of protein layers appeared to have a linear correlation (Fig. 3b). These observations suggest that surface zeta potentials may be a predictive indicator of the affinity of the surface for NP deposition but not necessarily of the total deposition extent. This phenomenon may be in part due to the longer range of electrostatic attractive forces that have a more direct impact on the initial NP deposition rate, whereas the total NP deposition is influenced not just by electrostatic but also shorter-range hydrophobic and other forces.14 Additionally, we have previously demonstrated that the overall surface zeta potential may not be capturing the necessary details for the likelihood of NP deposition onto polysaccharide-coated planar surfaces; instead, it was necessary to examine the surface potential heterogeneity of the organic layer.22
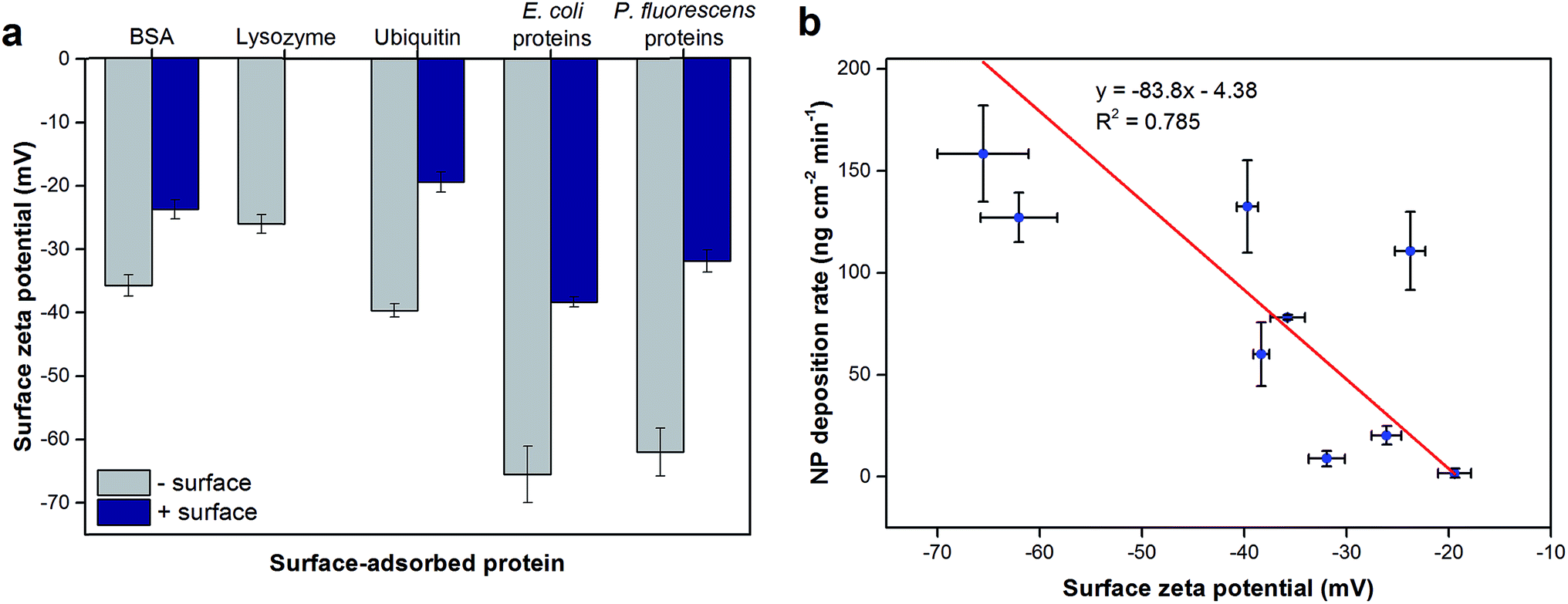 | ||
| Fig. 3 (a) Surface zeta potentials of model and extracted proteins adsorbed on negatively- or positively-charged sensor surfaces. (b) The relationship between surface zeta potentials of protein-coated surfaces and NP deposition rates. Zeta potentials were calculated based on streaming potential measurements in 10 mM NaCl (pH 5.7). Lysozyme only adsorbed onto negatively-charged sensor surfaces; as such, no data were collected for positively-charged surfaces. Linear regression analysis was performed on (b) and the resulting curve is shown. Error bars indicate the standard deviations of at least triplicate measurements. | ||
Similarly, we examined the surface potential variability of the protein layers by KPFM (Fig. S5 and S6†). Lysozyme and ubiquitin exhibited larger charge heterogeneity regardless of the bottom surface compared to BSA (p < 0.01). The charge and chemical nature of the bottom surface did not appear to affect the surface charge heterogeneity for all protein layers (p > 0.05) except for P. fluorescens protein extracts in which the negatively-charged bare sensor resulted in a greater variability (p = 0.0019). As shown in the representative KPFM images of the topmost surface of the protein layers (Fig. S5†), these surface charge heterogeneities largely result from small patches of more negative or positive charges (compared to the surrounding areas) that are tens to hundreds of nm in diameter. It is important to note that while there were some observable differences in the patches in the KPFM images, there was no clear trend other than the quantitative heterogeneity reported as the root mean squares of potentials across an area (Fig. S6†). Such variability in surface charge across the protein layer may be contributing to the observed discrepancies between the surface zeta potentials (Fig. 3a) and hematite NP deposition characteristics (Fig. 1). In addition, these surface charge results indicate that electrostatic interactions are not the only dominant interaction force between hematite NPs and protein layers.
To probe the hydrophobicity of each protein layer surface, contact angles as a measure of surface wettability of the surface-adsorbed protein layers are shown in Fig. 4a. While the contact angles indicated that all protein layers had hydrophilic surfaces (<90°), some layers were less hydrophilic due to different bottom surfaces or the identities of proteins. The bottom surface significantly changed the contact angles of the resulting protein layer for BSA and ubiquitin (p < 0.05) in which the protein layers adsorbed onto positively-charged planar surfaces appeared to be less hydrophilic. This observation did not apply to bacterial protein extracts likely because these protein layers are comprised of not a single type of protein but multiple proteins of various size and physicochemical nature, thus making such trends less observable. Furthermore, the contact angles of lysozyme layers were comparable to BSA and ubiquitin layers adsorbed on positively-charged bottom surfaces (p > 0.05). In addition, TOF SIMS was used to determine the chemical profile of the top surface of the protein layers (2–3 nm from the top) that is available for NP deposition. The ratios between amino acids with hydrophobic side chains to those with hydrophilic side chains found at the topmost surface of the protein layers were calculated as described by Baio et al.29 and are reported in Fig. 4b. These theoretical calculations for hydrophobicity showed similar trends as the contact angle measurements for most proteins tested and supported the overall trends of surface hydrophilicity of each protein layer as discussed above. The differences observed between the results shown in Fig. 3a and b were likely due to differences in the techniques with contact angle measurements conducted under ambient conditions whereas TOF SIMS analysis was done under vacuum. However, the contact angle values of the protein layers appeared to have no direct correlation with hematite NP deposition extents and rates (Fig. S7†).
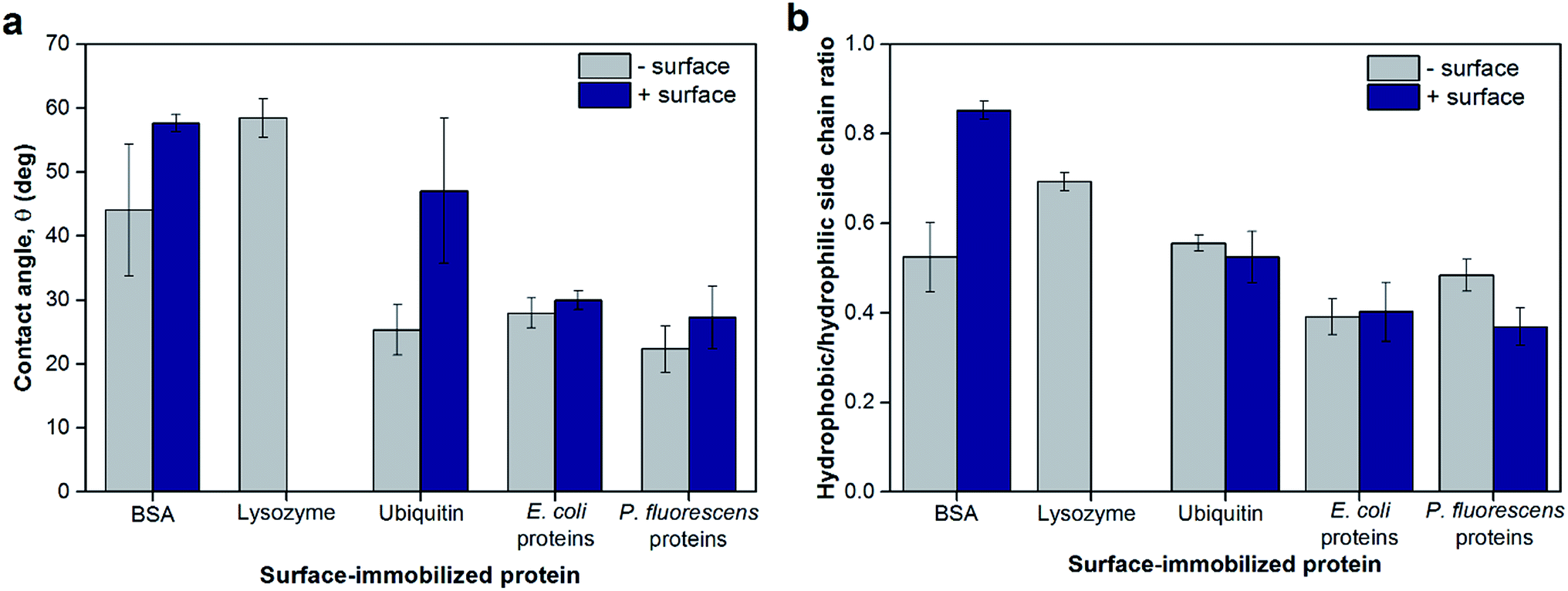 | ||
| Fig. 4 (a) Surface wettability of model and extracted proteins adsorbed on negatively- or positively-charged sensor surfaces measured as contact angles of water droplets. Lysozyme only adsorbed onto negatively-charged sensor surfaces; as such, no data were collected for positively-charged surfaces. (b) The ratios of hydrophobic-to-hydrophilic side chains at the topmost layer of surface-adsorbed proteins as determined by TOF SIMS. As described by Baio et al.,29 Ile/Leu, Met, Phe, and Val were designated as amino acids with hydrophobic side chains, whereas Arg, Asn, Gln, Glu, and His were designated as amino acids with hydrophilic side chains. Error bars indicate the standard deviations of at least triplicate measurements. | ||
Interaction mechanisms
The aforementioned differences in physicochemical characteristics of the adsorbed protein layer strongly suggest that the conformation and/or orientation of the surface-adsorbed model proteins is significantly different depending on the bottom surface (as depicted in Fig. 5), which is in agreement with previous studies.28,30–33 It is important to note that while the different orientations are due in part to the different surface charge of the QCM sensor, the slightly hydrophobic nature of the positively-charged PLL layer34 may have also contributed to such protein orientations.35 The differences in conformation and orientation as well as the identities of surface-adsorbed proteins result in different amino acid chemistry, charges, and hydrophobicity at the topmost surface available for interaction, thus greatly impacting the subsequent NP deposition characteristics (Fig. 5). Further investigation into the molecular mechanisms into the orientation/conformation of the planar surface-adsorbed proteins as well as the specific interaction forces governing NP deposition behavior onto such surface-adsorbed proteins are warranted. In addition, while this phenomenon was not directly observed in our QCM experiments, some of the proteins adsorbed onto the sensor surface may have desorbed and subsequently interacted with NPs in the bulk suspension to form a protein corona on the NP surface. These protein-coated NPs may have different deposition behaviors onto a protein-coated bulk planar surface wherein protein–protein interactions become one of the major forces at play.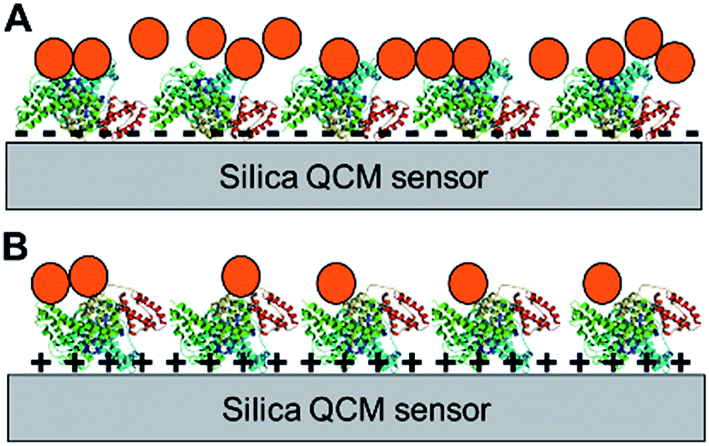 | ||
| Fig. 5 Examples of different protein orientations on planar surfaces and the resulting NP deposition characteristics. Potential orientations of BSA adsorbed on a negatively-charged bare silica sensor (A) or a positively-charged surface (B) and the resulting NP (orange circles) deposition onto the protein layers. The objects shown are not to scale. | ||
NP deposition onto bulk surfaces coated with organics is a complex interaction involving various attractive and repulsive forces. The results reported herein indicate that the interactions between NPs and protein-coated surfaces are similarly complex and involve both electrostatic and hydrophobic interactions, which are also considered the two major driving forces involved in protein corona formation on NPs.36 The interplay of electrostatic and hydrophobic interactions has been highlighted in several studies focused on NP–protein interactions in bulk suspension.36–38 However, unlike proteins in suspension, surface-adsorbed proteins lack the freedom of movement and therefore, the surface physicochemical features that are predetermined by the orientation and conformation of the bound proteins directly impacts NP deposition. Our results suggest that while electrostatic interactions were a major force, the difference in hydrophilicity of the surfaces may be an additional important factor that affected the deposition characteristics of hydrophilic hematite NPs. For example, the less hydrophilic nature of the protein layers of lysozyme as well as BSA and ubiquitin adsorbed on positively-charged surfaces (Fig. 4a) combined with the relatively small negative surface zeta potentials (Fig. 3a) resulted in very small NP deposition extents as seen in Fig. 1a. In these cases, repulsion of hydrophilic hematite NPs from adsorbing onto the less hydrophilic protein surfaces may have lowered the overall deposition extent than expected if the interactions were dominated by just electrostatic forces. Interestingly, synergistic effects between electrostatic and hydrophobic interaction forces have been reported for amino acid adsorption onto cation-exchange resins; specifically, weakly attractive hydrophobic interactions increased adsorption even when there was little electrostatic attraction.39 Our observations suggest that hydrophobic repulsion may lower the effect of weak electrostatic attraction, indicating that various combined effects of weak hydrophobic and electrostatic forces can be expected. However, not surprisingly, such combined effects do not linearly apply to all protein surfaces. The opposite trend was observed for BSA and ubiquitin layers adsorbed on negatively-charged sensor surfaces, in which a less hydrophilic protein layer (BSA; Fig. 4) resulted in larger hematite NP deposition extents (Fig. 1a) compared to the more hydrophilic surface (ubiquitin). The initial NP deposition rates were greater for the more hydrophilic ubiquitin surface (Fig. 1b). In these cases, these BSA and ubiquitin surfaces had comparable negative zeta potentials but significantly different surface potential heterogeneity (p = 0.0036, Fig. S6†). As such, while the initial deposition rates were not observably impacted, the differences in hematite NP deposition extents may have been more heavily affected by the small-scale variability in surface potential in these situations. In fact, all interaction forces must be evaluated at the nanoscale whenever possible especially when assessing interfacial NP–protein interactions, as protein surfaces have different nano-sized patches of potential deposition sites for NPs with varying affinities.
Conclusions
In real biological or environmental systems, most surfaces (such as blood vessels and various implants and biomaterials,35,40,41 filtration membranes,42 and soil minerals43) are expected to be covered with complex mixtures of proteins and other organic molecules. While our results with the model protein layers suggest that different proteins can result in different surface characteristics and subsequent NP deposition, such differences were less observable for complex total protein mixtures extracted from E. coli and P. fluorescens. In fact, overall, the nature of the bottom surface to which protein adsorb had a greater influence on surface physicochemical parameters and, in turn, on the NP deposition characteristics than the identities of proteins present. It is important to note that NPs entering real systems will likely be covered by layers of proteins and other organic molecules before approaching a protein-coated surface. While such realistic situations must be studied further, this study points to the importance of NP interactions with proteins adsorbed on bulk surfaces as another mode of NP–protein interactions. Furthermore, we showed that protein orientation/conformation and the resulting characteristics of the topmost surface are critical in the assessment of NP deposition onto adsorbed proteins. Future studies are needed to investigate the molecular mechanisms of such interactions as well as the role of proteins in complex organic coatings of bulk surfaces to which NPs are exposed.Acknowledgements
This work was supported by the National Science Foundation (CHE 1213546 and CBET 1454443) and the C. Gus Glasscock, Jr Endowed Fund for Excellence in Environmental Science from Baylor University. We thank Andrew Madden for the generous donation of the hematite NPs used in this study, and Sven Behrens and Jamie Grunlan for providing instrument access.References
- I. Lynch and K. a Dawson, Nano Today, 2008, 3, 40–47 CrossRef CAS.
- M. Mahmoudi, I. Lynch, M. R. Ejtehadi, M. P. Monopoli, F. B. Bombelli and S. Laurent, Chem. Rev., 2011, 111, 5610–5637 CrossRef CAS PubMed.
- P. Del Pino, B. Pelaz, Q. Zhang, P. Maffre, G. U. Nienhaus, W. J. Parak, P. del Pino and P. D. U. Nienhaus, Mater. Horiz., 2014, 1, 301–313 RSC.
- S. R. Saptarshi, A. Duschl, A. L. Lopata, A. Duschl and A. L. Lopata, J. Nanobiotechnol., 2013, 11, 26 CrossRef CAS PubMed.
- M. P. Monopoli, F. B. Bombelli and K. A. Dawson, Nat. Nanotechnol., 2011, 6, 11–12 CrossRef CAS PubMed.
- M. Mahmoudi, S. E. Lohse, C. J. Murphy, A. Fathizadeh, A. Montazeri and K. S. Suslick, Nano Lett., 2014, 14, 6–12 CrossRef CAS PubMed.
- Y. Dudal and F. Gérard, Earth-Sci. Rev., 2004, 66, 199–216 CrossRef CAS.
- S. S. Branda, A. Vik, L. Friedman and R. Kolter, Trends Microbiol., 2005, 13, 20–26 CrossRef CAS PubMed.
- H. Flemming and J. Wingender, Nat. Rev. Microbiol., 2010, 8, 623–633 CAS.
- S. Kalasin and M. M. Santore, Colloids Surf., B, 2009, 73, 229–236 CrossRef CAS PubMed.
- Z. Adamczyk, M. Nattich, M. Wasilewska and M. Sadowska, J. Colloid Interface Sci., 2011, 356, 454–464 CrossRef CAS PubMed.
- Z. Shi, K. G. Neoh, E.-T. Kang, C. Poh and W. Wang, Tissue Eng., Part A, 2009, 15, 417–426 CrossRef CAS PubMed.
- O. Furman, S. Usenko and B. L. T. Lau, Environ. Sci. Technol., 2013, 47, 1349–1356 CAS.
- K. L. Chen and M. Elimelech, Environ. Sci. Technol., 2008, 42, 7607–7614 CrossRef CAS PubMed.
- J. Fabrega, J. C. Renshaw and J. R. Lead, Environ. Sci. Technol., 2009, 43, 9004–9009 CrossRef CAS PubMed.
- E. Sahle-Demessie and H. Tadesse, Surf. Sci., 2011, 605, 1177–1184 CrossRef CAS.
- J. Spence, S. Sadis, A. L. Haas and D. Finley, Mol. Cell. Biol., 1995, 15, 1265–1273 CrossRef CAS PubMed.
- R. L. Moritz and R. J. Simpson, Nat. Methods, 2005, 2, 863–873 CrossRef CAS PubMed.
- U. Schwertmann and R. M. Cornell, Iron Oxides in the Laboratory. Preparation and Characterization, Wiley-VCH, Weinheim, Germany, 2000 Search PubMed.
- J. M. Walker, The Protein Protocols Handbook, Humana Press, Totowa, NJ, 2002 Search PubMed.
- B. L. T. Lau, R. Huang and A. S. Madden, J. Nanopart. Res., 2013, 15(8), 1873–1882 CrossRef.
- K. Ikuma, A. S. Madden, A. W. Decho and B. L. T. Lau, Environ. Sci.: Nano, 2014, 1, 117 RSC.
- I. Reviakine, D. Johannsmann and R. P. Richter, Anal. Chem., 2011, 83, 8838–8848 CrossRef CAS PubMed.
- M. S. Wagner and D. G. Castner, Langmuir, 2001, 17, 4649–4660 CrossRef CAS.
- F. Fairbrother and H. Mastin, J. Chem. Soc., Trans., 1924, 125, 2319–2330 RSC.
- N. Reitzel, T. Hassenkam, K. Balashev, T. R. Jensen, P. B. Howes, K. Kjaer, A. Fechtenkotter, N. Tchebotareva, S. Ito, K. Mullen and T. Bjornholm, Chem.–Eur. J., 2001, 7, 4894–4901 CrossRef CAS.
- M. Madliger, M. Sander and R. P. Schwarzenbach, Environ. Sci. Technol., 2010, 44, 8877–8883 CrossRef CAS PubMed.
- R. A. Silva, M. D. Urzua, D. F. S. Petri and P. L. Dubin, Langmuir, 2010, 26, 14032–14038 CrossRef CAS PubMed.
- J. E. Baio, T. Weidner, G. Interlandi, C. Mendoza-Barrera, H. E. Canavan, R. Michel and D. G. Castner, J. Vac. Sci. Technol., B: Microelectron. Nanometer Struct., 2011, 29, 04D113 Search PubMed.
- K. Wadu-Mesthrige, N. A. Amro and G. Y. Liu, Scanning, 2000, 22, 380–388 CrossRef CAS PubMed.
- A. C. Pinho and A. P. Piedade, ACS Appl. Mater. Interfaces, 2013, 5, 8187–8194 CAS.
- H. Okusa, K. Kurihara and T. Kunitake, Langmuir, 1994, 10, 3577–3581 CrossRef CAS.
- A. Sethuraman, M. Han, R. S. Kane and G. Belfort, Langmuir, 2004, 20, 7779–7788 CrossRef CAS PubMed.
- A. Maruyama, T. Ishihara, J. S. Kim, S. Wan Kim and T. Akaike, Colloids Surf., A, 1999, 153, 439–443 CrossRef CAS.
- P. Roach, D. Farrar and C. C. Perry, J. Am. Chem. Soc., 2005, 8168–8173 CrossRef CAS PubMed.
- N. Welsch, Y. Lu, J. Dzubiella and M. Ballauff, Polymer, 2013, 54, 2835–2849 CrossRef CAS.
- F. Turci, E. Ghibaudi, M. Colonna, B. Boscolo, I. Fenoglio and B. Fubini, Langmuir, 2010, 26, 8336–8346 CrossRef CAS PubMed.
- T. Cedervall, I. Lynch, S. Lindman, T. Berggård, E. Thulin, H. Nilsson, K. A. Dawson and S. Linse, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 2050–2055 CrossRef CAS PubMed.
- S. Cheng, H. Yan and C. Zhao, J. Chromatogr. A, 2006, 1108, 43–49 CrossRef CAS PubMed.
- H. Nojiri, S. Nakahama, K. D. Park and S. W. Kim, J. Biomater. Sci., Polym. Ed., 1993, 4, 75–88 CrossRef.
- D. Deligianni, N. Katsala, S. Ladas, D. Sotiropoulou, J. Amedee and Y. Missirlis, Biomaterials, 2001, 22, 1241–1251 CrossRef CAS PubMed.
- K. Nakamura and K. Matsumoto, J. Membr. Sci., 2006, 280, 363–374 CrossRef CAS.
- M. H. Baron, M. Revault and H. Quiquampoix, in Spectroscopy of Biological Molecules: Modern Trends, ed. P. Carmona, R. Navarro and A. Hernanz, Springer Netherlands, Dordrecht, The Netherlands, 1997, pp. 503–504 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra13508k |
| This journal is © The Royal Society of Chemistry 2016 |