
Greenly synthesized graphene with l-glutathione-modified electrode and its application towards determination of rutin†
Abstract
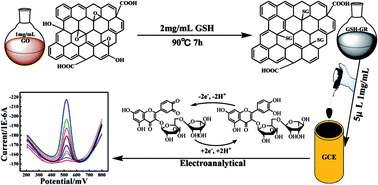
A simple, environmentally friendly and cost-effective approach for preparation of water-soluble functional graphene was proposed for constructing a voltammetric sensor platform. It was characterized by several techniques, including UV-vis spectroscopy, Fourier transform infrared spectroscopy, transmission electron microscopy (TEM) and X-ray diffraction (XRD). This voltammetric sensor showed strong accumulation ability and excellent voltammetric response for rutin. The electrochemical behavior of rutin was investigated systematically in 0.1 mol L−1 phosphate buffer solution (PBS 3.0). Under optimum conditions by square wave voltammetry (SWV), a good linear relationship was obtained between peak currents and rutin concentrations in the wider range of 5 × 10−9 to 1 × 10−6 mol L−1 with a detection limit of 7 × 10−10 mol L−1. In addition, the proposed rutin sensor was successfully applied to test in real samples with good results.
 Please wait while we load your content...
Please wait while we load your content...

