
The redox mechanism of NpVI with hydrazine: a DFT study†
Zhong-Ping Cheng‡
ab,
Qun-Yan Wu‡a,
Yun-Hai Liub,
Jian-Hui Lana,
Cong-Zhi Wanga,
Zhi-Fang Chaiac and
Wei-Qun Shi*a
aLaboratory of Nuclear Energy Chemistry, Key Laboratory for Biomedical Effects of Nanomaterials and Nanosafety, Institute of High Energy Physics, Chinese Academy of Sciences, Beijing, 100049, China. E-mail: shiwq@ihep.ac.cn
bSchool of Chemistry, Biological and Materials Sciences, East China University of Technology, Nanchang 330013, China
cSchool of Radiological and Interdisciplinary Sciences (RAD-X), Collaborative Innovation Center of Radiation Medicine of Jiangsu Higher Education Institutions, Soochow University, Suzhou 215123, China
First published on 10th November 2016
Abstract
Valence state control and adjustment of neptunium in spent fuel reprocessing is very important for improving the separation efficiency of U/Np and Np/Pu. Hydrazine and its derivatives have been experimentally demonstrated to be effective in the reduction of NpVI to NpV. In this work, hydrazine was used as a representative reductant and the reduction mechanisms of NpVI induced by hydrazine were investigated using density functional theory (DFT) calculations. Three reaction pathways were taken into account and characterized by gradually transferring a hydrogen atom from N2H4 to the “yl”-oxygen of [NpVIO2(H2O)5]2+ followed by the valence state adjustment from NpVI to NpV. The calculated results of the potential energy profiles (PEPs) revealed that Pathway I should be the most likely to occur as the process of forming ˙N2H3 is considered to be the rate-determining step with the highest energy barrier of 32.02 kcal mol−1, which is in favor of the experimental results. Pathway II hardly occurs and Pathway III probably occurs. The bonding evolution, along with the reaction pathways, was explored through natural bond orbitals (NBOs), quantum theory of atoms-in-molecules (QTAIM) and electron localization function (ELF) analyses. This work can shed light on the understanding of redox mechanisms of NpVI with N2H4 and its derivatives and help further attempts to design more efficient reductants for the separation of U/Np and Np/Pu in spent nuclear fuel reprocessing in the near future.
Introduction
The treatment of neptunium in the nuclear fuel cycle is of significant concern since 237Np is present in non-negligible quantities in spent nuclear fuels and can migrate into the environment under specific conditions due to the stability of the pentavalent neptunyl ion (NpO2+).1 Recovery and recycling of Np, either to diminish the long term radiotoxicity or to reduce the heat burden on the geological repository, is therefore under investigation in most nuclear power countries.2 It is known that in the so-called Plutonium URanium Extraction (PUREX) process,3 which is used for the commercial scale reprocessing of spent nuclear fuels, it is probable to recover and recycle Np through the valence state control of Np, as Np can be routed with Pu or U streams in the partition contactor, and disparate oxidation states of Np such as NpIV, NpV and NpVI can emerge in this process. It is also well known that the extraction behaviors of neptunium towards TBP in nitric acid medium vary in different oxidation states;4 NpVI was readily extractable, NpIV was relatively poor extractable, and NpV was barely extractable. However, the three oxidation states of Np can interconvert easily by various redox reactions under the conditions of solvent extraction-based spent nuclear fuel reprocessing.So far, two approaches have been used to control and adjust the oxidation states of Np.5,6 One was through in situ redox reaction based on photochemical induction7–9 or electrochemistry.10–12 For instance, Wada and his co-workers adjusted the oxidation states of Np to NpV in nitric acid medium by a photochemical method.8 Kim et al. studied the electrochemical behavior of NpO22+ cation on platinum and glassy carbon electrodes,12 where the NpO22+ cation was reduced to NpV quasi-reversibly. The other was via ex situ redox reaction based on a specific redox reagent. For example, Bibler et al. employed ferrous sulfamate as a reductant for reducing NpVI/V to NpIV in the PUREX process.13 However, these redox reagents can introduce extra large amounts of salts to the process and will increase the amount of wastes. To avoid these defects, some new salt-free reductants, such as aldehydes,14,15 hydroxylamine derivatives16–18 and hydrazine derivatives19–27 were developed and confirmed to be effective reductants. For instance, Koltunov and his co-workers investigated the reduction kinetics of NpVI with hydrazine derivatives.25 It was found that the reaction rate constants of NpVI varied with respect to different substituent of the hydrazine derivatives. Recently, Ban et al. investigated the reduction kinetic of NpVI with N,N-dimethylhydrazine in spent fuel reprocessing,19 in which NpVI was quickly reduced to NpV. Nevertheless, previous works about NpVI reduction only focused on experimental researches. Reduction mechanisms were still unclear. In addition, to the best of our knowledge, theoretical investigations have been rarely addressed on this issue. Therefore, it was extremely necessary and meaningful to investigate the reduction mechanism of NpVI by theoretical calculations, which can give reasonable description about geometries and properties of the actinide complexes.28–31
In this work, the three reduction mechanisms of NpVI with N2H4 were carried out based on the results of DFT (density functional theory) calculations. And we investigated systematically the geometries and stabilities of stationary points of potential energy profiles (PEPs). The solvent effect had been taken into account. The optimal reaction pathway of NpVI with N2H4 was obtained by comparing the energy barriers. This work can provide theoretical insights for having a command of the reduction mechanisms of NpVI with N2H4 and its derivatives, and it is expected that more suitable reagents can be found to improve the separation efficiency of U/Np and Np/Pu in spent nuclear fuel reprocessing in the future.
Computational details
All quantum chemical calculations were performed using the hybrid B3LYP functional32–37 within the Gaussian 09 program package.38 For geometry optimizations, the quasi-relativistic small-core pseudopotential ECP60MWB coupled with ECP60MWB-SEG valence basis sets were employed for neptunium,39–41 while the 6-31g* basis set was used for the other light atoms H, N, O. The quasi-relativistic small-core pseudopotential substituted 60 core electrons for neptunium, whereas the remaining 33 electrons were represented by the associated valence basis set. A small-core ECP was prevalently considered to obtain more accurate results compared to a large ECP.42–44 Geometry optimizations of all the species were performed without symmetry restrictions in the aqueous phase at the B3LYP/ECP60MWB/6-31g* level of theory by using COSMO (conductor-like screening model).45,46 Computations of open-shell systems were performed with spin-unrestricted methods. The minimal (without imaginary frequency) and transition states (with one imaginary frequency) of complexes in the three pathways were confirmed with harmonic frequency calculations and vibrational mode analyses which were performed by the GaussView program.47 The intrinsic reaction coordinate (IRC)48,49 calculations were carried out to confirm that the transition states connected reactants and products. The PEPs of three reaction pathways of [NpVIO2(H2O)5]2+ with N2H4 were obtained by calculating the relative bond energies of each species with respect to the ground-state reactant.Recently, there are significant developments in understanding the accuracy and applicability of the DFT methods, especially for the chemistry of actinide complexes. Therefore, to achieve the accurate description of neptunium complexes, the influence of different functionals (B3LYP, PBE, BP86 and M06) and basis sets (6-31g*, 6-311+g* and 6-311++g**) on the geometries of [NpVIO2(H2O)5]2+ and [NpVO2(H2O)5]2+ have been investigated in this work. The more complete report of computational results is provided in ESI.† It is shown from Table S1† that the B3LYP functional and 6-31g* basis set for this work are reasonable and acceptable according to the comparison between the predicted bond lengths and available experimental data. In addition, for the neptunium atom with f-electron, effects of spin–orbital (SO) coupling are expected to be huge.50 It has also been shown that SO coupling can affect reaction barrier dramatically.51 Therefore, further computations were undertaken for sake of evaluating the effects of spin–orbital coupling on the PEPs. In particular, the single-point computations were carried out on the B3LYP-optimized structures using the zero-order regular approximation (ZORA) associated with the spin–orbital correction (SO-ZORA). The SO-ZORA approximations were employed together with the BP86 functional and triple-ζ basis sets (TZP) without frozen core in the implementation of Amsterdam density functional (ADF 2013.01) package.52,53 PEPs of the three pathways are obtained at the BP86-SO-ZORA/TZP level of theory.
The bonding properties of all the minima and transition states involved in the reaction pathways were explored with the electron localization function (ELF)54–56 analysis and the quantum theory of atoms-in-molecules (QTAIM) method57–59 by using Multiwfn program.57 In particular, we explored the properties of the (3, −1) bond critical points (BCPs) in the gradient field of the electron density. Lastly, the Wiberg bond index (WBI)60 and Mayer bond order61 of the related bonds were obtained by NBO (natural bonding orbital) analysis.
The abbreviations used in this work are as follows: (i) all species are denoted as icNM, tsNM and intNM (ic = initial complex, ts = transition state, int = intermediate, M = I, II, III and N = 1 to 4); (iii) four hydrogen and two nitrogen atoms of N2H4 are labeled as H1, H2, H3, H4, N1 and N2 respectively.
Results and discussion
According to the experimental results of Koltunov and his co-workers,25 the total reaction equation of [NpVIO2(H2O)5]2+ with N2H4 in the aqueous phase can be depicted as follows:| 4[NpVIO2(H2O)5]2+ + N2H4 → 4[NpVO2(H2O)5]+ + N2 + 4H+ | (1) |
It has been known to be a consecutive reaction, hence, three probable pathways were designed to understand their reduction mechanisms. Because each stage of the same pathway is similar, here, we took the first stage of three pathways as an example, as shown in Scheme 1. It can be observed from Scheme 1 that the formation of int1 is the same process for the three pathways.
 | ||
| Scheme 1 The redox of [NpVIO2(H2O)5]2+ with N2H4 in the first stage of Pathways I, II and III. | ||
(1) Pathway I via a free radical mechanism: from Scheme 1, it can be clearly seen that the H1 of ic1 is transferred to the “yl”-oxygen (Oyl) of [NpVIO2(H2O)5]2+ through N–H1 bond dissociation, forming the int1. After int1 direct dissociation, the products such as [NpVO2(H2O)5]+ and ˙N2H3 are obtained. The remaining hydrogen atoms are gradually transferred to the Oyl atom of [NpVIO2(H2O)5]2+ in the same way. Each stage of Pathway I can be expressed as following:
| [NpVIO2(H2O)5]2+ + N2Hx → [NpVO2(H2O)5]+ + N2Hx−1 + 4H+ | (2) |
(2) Pathway II via a free radical and ion mechanism: after forming int1, H atom of equatorial water molecule of int1 is close to N1 atom of int1 to form int1II accompanying with the Oeq.–Heq. bond dissociation. After that, H1 atom of int1II is transferred to the hydroxyl of equatorial plane of int1II to obtain [NpVO2(H2O)5]+ and ˙N2H4+. The other stages of Pathway II are similar to the first stage of Pathway I. Each stage of Pathway II can be expressed as following:
| [NpVIO2(H2O)5]2+ + N2H4 → [NpVO2(H2O)5]+ + N2H4+ | (3) |
| [NpVIO2(H2O)5]2+ + N2Hx+ → [NpVO2(H2O)5]+ + N2Hy+ + (x − y)H+ | (4) |
(3) Pathway III via a free radical and disproportionation mechanism: in the first stage, both H1 and H2 are gradually transferred to Oyl atom and the products such as [NpIVO2H2(H2O)5]2+ and N2H2 are obtained. Similarly, H3 and H4 are subsequently transferred to get [NpIVO2H2(H2O)5]2+ and N2. Eventually, [NpIVO2H2(H2O)5]2+ reacts with [NpVIO2(H2O)5]2+ to obtain [NpVO2(H2O)5]+ by a disproportionation mechanism. Each stage of Pathway III can be expressed as following:
| [NpVIO2(H2O)5]2+ + N2Hx → [NpIVO2H2(H2O)5]2+ + N2Hx−2 | (5) |
| [NpVIO2(H2O)5]2+ + [NpIVO2H2(H2O)5]2+ → 2[NpVO2(H2O)5]+ + 2H+ | (6) |
Pathway I via a free radical mechanism
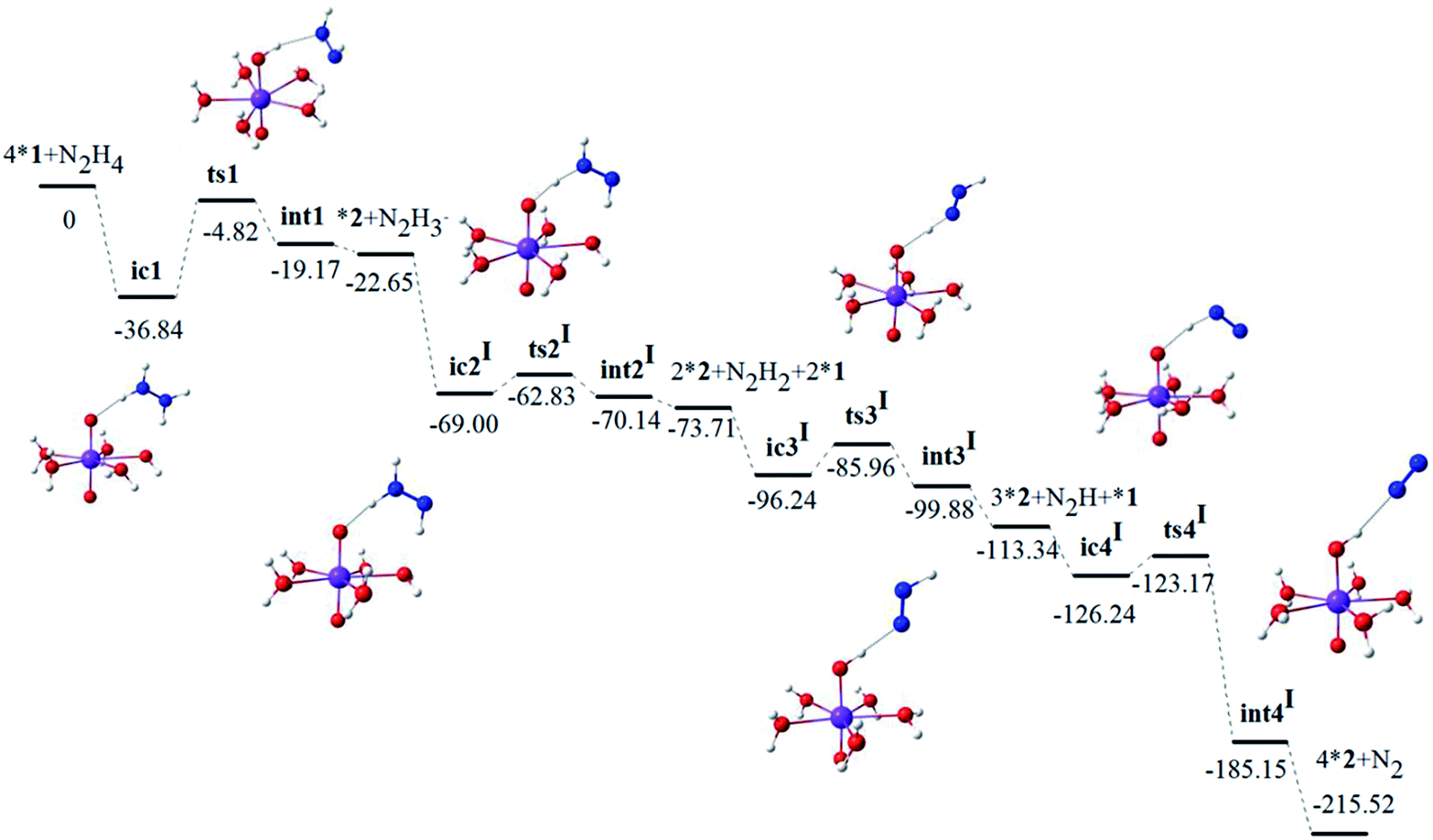 | ||
| Fig. 1 Potential energy profile (ΔE, kcal mol−1) of Pathways I computed at the BP86-SO-ZORA/TZP level of theory. The [NpVIO2(H2O)5]2+ and [NpVO2(H2O)5]+ were denoted as *1 and *2 respectively. | ||
The optimized geometries and related bond lengths of all species in the Pathway I are presented in the ESI, Fig. S1.† As shown in Fig. S1(a), ESI† from ic1 to ts1 and int1, the N–H1/Np–Oyl bond length increases from 1.031/1.825 Å to 1.432/1.956 Å and 2.041/1.985 Å, respectively, while Oyl–H1 bond length decreases from 1.853 Å to 1.429 Å and 0.997 Å, indicating N–H1 bond dissociation and Oyl–H1 bond formation. Like the first stage, the trends of related N–H, Np–Oyl and Oyl–H bond lengths from ic to ts and int for the other stages of Pathway I as presented in Fig. S1(b)–(d), ESI† are similar.
It can be known that the Wiberg bond index between atom A and B is defined as
 | (7) |
And Mayer bond order between atom A and B is defined as
 | (8) |
| WBI | Mayer bond order | |||||
|---|---|---|---|---|---|---|
| Oyl–Ha | Np–Oyl | N–Ha | Oyl–Ha | Np–Oyl | N–Ha | |
| a H represents H1–H4 in N2H4 corresponding to four stages of Pathway I. | ||||||
| ic1 | — | 1.924 | 0.729 | — | 1.940 | 0.641 |
| ts1 | 0.298 | 1.584 | 0.409 | 0.338 | 1.557 | 0.385 |
| int1 | 0.620 | 1.281 | — | 0.623 | 1.253 | — |
| ic2I | 0.091 | 1.868 | 0.664 | 0.169 | 1.844 | 0.553 |
| ts2I | 0.217 | 1.736 | 0.508 | 0.249 | 1.694 | 0.446 |
| int2I | 0.509 | 1.371 | 0.188 | 0.491 | 1.360 | 0.196 |
| ic3I | — | 2.233 | 0.801 | — | 2.168 | 0.791 |
| ts3I | 0.186 | 1.678 | 0.545 | 0.278 | 1.630 | 0.407 |
| int3I | 0.587 | 1.314 | — | 0.566 | 1.304 | — |
| ic4I | — | 2.210 | 0.726 | — | 2.116 | 0.675 |
| ts4I | 0.093 | 2.080 | 0.607 | 0.121 | 1.954 | 0.548 |
| int4I | 0.638 | 1.277 | — | 0.609 | 1.262 | — |
Certainly, the process of the bonding evolution is also supported by the QTAIM analysis. It is well worth mentioning that QTAIM is a ubiquitous approach for studying the properties and evolution of chemical bonds.63–65 In principle, at BCP the electron density ρ(r) > 0.20 au and Laplacian ∇2ρ(r) < 0 present a covalent bond (shared shell interaction), while ρ(r) < 0.10 au and Laplacian ∇2ρ(r) > 0 indicate an ionic bond (closed shell interaction). The total energy density H(r) at the BCP is negative for interactions with significant sharing of electrons, which magnitude can reflect the “covalence” of the interaction.66 However, it is unanticipated that the above criteria of QTAIM analysis are not available for strong polar covalent bond. Therefore, the other criteria that ρ(r) > 0.20 au and H(r) < 0 present a covalent bond (shared shell interaction), while ρ(r) < 0.10 au and H(r) > 0 indicate an ionic bond (closed shell interaction) are employed.66,67 The ρ(r), ∇2ρ(r) and H(r) at the BCPs are listed in Table 2. It is shown that the ρ(r) at Oyl–H BCP for each intermediate are over 0.2 au with negative ∇2ρ(r). Moreover, with reaction proceeding from initial complex to intermediate for each stage, the ρ(r) at N–H BCP gradually decreases. From these results, we conclude that N–H bond is dissociated and Oyl–H bond is formed in every stage of Pathway I. In addition, Np–Oyl bond is a strong polar covalent bond with the value of larger ρ(r) and negative H(r) for each stationary point. And the strength of Np–Oyl gradually weakens with ρ(r) decreasing from initial complex to intermediate for each stage, which indirectly reflects the formation of Oyl–H bond. Besides, we also perform the ELF analysis to further confirm the characteristic of bonding interaction. It has been discovered that the ELF analysis can afford effective information on chemical bonds.68–70 In generally, ELF falls within the scope of 0 and 1, ELF = 1 suggests the faultless electron localization, whereas ELF = 0 reveals the perfect electron delocalization.54,55 The ELF value is closer to 1 suggesting stronger covalence of a chemical bond. Fig. 2 presents two-dimensional colored ELF pictures of all species on Np–Oyl–N plane. It is obviously known that from initial complex to intermediate for each stage, the ELF value of N–H bond gradually decreases, and that of Oyl–H bond increases, suggesting the dissociation of N–H bonds and formation of Oyl–H bonds.
| Oyl–Ha | N–Ha | Np–Oyl | |||||||
|---|---|---|---|---|---|---|---|---|---|
| ρ(r) | ∇2ρ(r) | H(r) | ρ(r) | ∇2ρ(r) | H(r) | ρ(r) | ∇2ρ(r) | H(r) | |
| a H represents H1–H4 in N2H4 corresponding to four stages of Pathway I. | |||||||||
| ic1 | 0.030 | 0.104 | 0.001 | 0.320 | −1.782 | −0.486 | 0.261 | 0.263 | −0.206 |
| ts1 | 0.094 | 0.073 | −0.028 | 0.108 | −0.056 | −0.048 | 0.191 | 0.335 | −0.102 |
| int1 | 0.301 | −1.520 | −0.434 | — | — | — | 0.171 | 0.367 | −0.008 |
| ic2I | 0.047 | 0.149 | −0.001 | 0.287 | −1.416 | −0.389 | 0.256 | 0.291 | −0.200 |
| ts2I | 0.082 | 0.141 | −0.018 | 0.154 | −0.364 | −0.125 | 0.256 | 0.323 | −0.199 |
| int2I | 0.252 | −1.044 | −0.326 | 0.067 | 0.136 | −0.010 | 0.186 | 0.379 | −0.095 |
| ic3I | — | — | — | 0.328 | −1.673 | −0.457 | 0.326 | 0.216 | −0.323 |
| ts3I | 0.057 | 0.114 | −0.005 | 0.188 | −0.700 | −0.210 | 0.188 | 0.350 | −0.098 |
| int3I | 0.294 | −1.450 | −0.420 | 0.038 | 0.109 | −0.001 | 0.178 | 0.367 | −0.087 |
| ic4I | 0.014 | 0.046 | 0.001 | 0.294 | −1.430 | −0.397 | 0.325 | 0.200 | −0.324 |
| ts4I | 0.048 | 0.136 | −0.002 | 0.186 | −0.544 | −0.171 | 0.321 | 0.224 | −0.317 |
| int4I | 0.314 | −1.660 | −0.467 | 0.019 | 0.067 | 0.002 | 0.170 | 0.378 | −0.077 |
 | ||
| Fig. 2 Two dimensioned ELF contours of all species on the Np–Oyl–N plane for four stages of Pathway I. | ||
Pathway II via a free radical and ion mechanism
 | ||
| Fig. 3 Potential energy profile (ΔE, kcal mol−1) of Pathways II computed at the BP86-SO-ZORA/TZP level of theory. The [NpVIO2(H2O)5]2+ and [NpVO2(H2O)5]+ were denoted as *1 and *2 respectively. | ||
| WBI | Mayer bond order | |||||
|---|---|---|---|---|---|---|
| Oyl–Ha | Np–Oyl | N–Ha | Oyl–Ha | Np–Oyl | N–Ha | |
| a H represents H2–H4 in N2H4 corresponding to stages 2, 3 and 4 of Pathway II.b H and O represent the Heq.–Oeq. of H2O in the equatorial plane corresponding to stage 1 of Pathway II. | ||||||
| int1 | 0.536b | — | 0.178b | 0.526b | — | 0.209b |
| ts1II | 0.361b | — | 0.386b | 0.375b | — | 0.324b |
| int1II | 0.104b | — | 0.678b | 0.198b | — | 0.580b |
| ic2II | — | 2.201 | 0.761 | — | 2.096 | 0.699 |
| ts2II | 0.138 | 2.058 | 0.591 | 0.143 | 1.925 | 0.600 |
| int2II | 0.648 | 1.276 | — | 0.683 | 1.224 | — |
| ic3II | — | 2.186 | 0.728 | — | 2.068 | 0.664 |
| ts3II | 0.145 | 2.034 | 0.540 | 0.163 | 1.888 | 0.532 |
| int3II | 0.650 | 1.262 | — | 0.685 | 1.208 | — |
| ic4II | — | 2.237 | 0.709 | — | 2.177 | 0.677 |
| ts4II | 0.148 | 2.010 | 0.471 | 0.195 | 1.856 | 0.415 |
| int4II | 0.652 | 1.263 | — | 0.680 | 1.202 | — |
As expected, these bonding interactions are also confirmed by QTAIM and ELF analyses. It can be known from Table S2, ESI† that ρ(r) at N–Heq. BCP for int1II is 0.288 au with negative ∇2ρ(r), revealing the H transfer from int1 to int1II. In stages 2, 3 and 4, the trends of ρ(r) at N–H, Np–Oyl and Oyl–H BCPs for Pathway II are similar to that of Pathway I. In addition, the two-dimensional colored ELF pictures of all species on Np–Oeq.–N and Np–Oyl–N planes are provided in Fig. S3 ESI.† The ELF value of Oeq.–Heq. decreases and that of N–Heq. increases, revealing the transfer of H from int1 to int1II. Moreover, in stages 2, 3 and 4, the ELF results of N–H and Oyl–H bonds for Pathway II are in analogy with those for Pathway I. These results suggest that the N–H bond is dissociated and Oyl–H bond is formed with the reaction proceeding from initial complex to intermediate for each stage of Pathway II.
Pathway III via a free radical and disproportionation mechanism
 | ||
| Fig. 4 PEP (ΔE, kcal mol−1) of stages 1 and 2 of Pathway III computed at the BP86-SO-ZORA/TZP level of theory. The [NpVIO2(H2O)5]2+ and [NpIVO2H2(H2O)5]2+ were denoted as *1 and *3 respectively. | ||
Fig. S4, ESI† shows structures and related bond lengths of all species in the first and second stages of Pathway III. As seen from Fig. S4(a)† and 4, the Oyl–H2 bond length decreases from 1.466 Å (ts1III) to 1.008 Å (int1III), while the N–H2/Np–Oyl bond length increases from 1.019/1.985 Å of int1 to 1.763/2.461 Å of int1III, which suggests H2 is successfully transferred to Oyl atom. The transfer of H4 to Oyl atom as shown in Fig. S4(b)† and 4 is similar to that of H2.
The three pathways with different mechanisms for reduction of NpVI with hydrazine molecule are proposed, and the feasibilities of three pathways are testified by DFT computation. According to the calculated PEPs, it can be inferred that the Pathway I is the most likely to occur, the Pathway II is hardly realized and Pathway III is probably taken place through comparing the energy barriers and the stability of species. The reason of high energy barrier in the Pathway II is likely to be that the nitrogen atom electron density of ˙N2H4+ is smaller together with the smaller electronegative of nitrogen atom compared to those of ˙N2H3, resulting in the weaker polarization of N–H bond that makes the dissociation of N–H bond become more difficult. In the Pathway III, the energy barriers of the second and fourth stages are relative higher than those of Pathway I. It is probably that the activity of neptunyl ion is so small that the first H atom transfer is relative easy, but successive transferring the second H atom becomes difficult. In addition, the nature of NpVI reduction is gradually transferring hydrogen atom from N2H4 to “yl”-oxygen of [NpVIO2(H2O)5]2+, which is embodied by the analyses of bond length, bond order, QTAIM and ELF. All the calculated results suggest that the N–H bond gradually dissociated and Oyl–H bond gradually formed with the reaction proceeding from initial complex to intermediate for each stage.
Conclusions
In this work, the redox mechanisms of NpVI with N2H4 in aqueous solution were investigated based on the difficult control and adjustment of neptunium valence state in spent fuel reprocessing. The three reaction pathways with free radical, free radical and ion, as well as free radical and disproportionation mechanism were proposed. And it is supposed that the redox nature of [NpVIO2(H2O)5]2+ with N2H4 was the gradual transfer of hydrogen atoms along with the dissociation of N–H bonds and formation of Oyl–H bonds. To verify the pathway reasonability, we have performed a series of theoretical calculations. All the structures of species of the three pathways are optimized at the B3LYP/ECP60MWB/6-31g* level of theory. Consideration the importance of spin–orbital coupling of neptunium atom, we performed the single-point computation based on the B3LYP-optimized structures at the BP86-SO-ZORA/TZP level of theory. The PEPs of three pathways were depicted based on the relative bond energies of each species with respect to the ground-state reactant. The results show that the Pathway I is the most reasonable among the three ones. Moreover, the stage of forming ˙N2H3 of Pathway I has the highest energy barrier of 32.02 kcal mol−1 and is the rate-determining step, which is in accordance with the experimental observation.25 In addition, in order to prove the redox nature, the analyses of bond length, the WBIs and Mayer bond orders, QTAIM and ELF were provided for whole reaction process. The calculated results explicitly explained the redox nature with the transferring H atoms step by step. In summary, this work has been provided the understanding of reduction mechanisms of NpVI with N2H4 and its derivatives. It is expected this work can provide theoretical basis on designing more efficient reductants for the separation of U/Np and Np/Pu in spent nuclear fuel reprocessing.Acknowledgements
This work was supported by the Natural Science Foundation of China (Grant No. 21477130, 91426302, 91326202) and the “Strategic Priority Research Program” of the Chinese Academy of Sciences (Grant No. XDA030104). The results describle in this work were obtained on the ScGrid of Supercomputing Center, Computer Network Information Center of Chinese Academy of Sciences.References
- R. C. Thompson, Radiat. Res., 1982, 90, 1–32 CrossRef CAS PubMed.
- C. Madic, M. Lecomte, P. Baron and B. Boullis, C. R. Phys., 2002, 3, 797–811 CrossRef CAS.
- R. S. Herbst, P. Baron and M. Nilsson, in Advanced Separation Techniques for Nuclear Fuel Reprocessing and Radioactive Waste Treatment, Woodhead Publishing, 2011, pp. 141–175 Search PubMed.
- J. C. Hindman, L. B. Magnusson and T. J. LaChapelle, J. Am. Chem. Soc., 1949, 71, 687–693 CrossRef CAS.
- R. J. Taylor, I. S. Denniss and A. L. Wallwork, Nucl. Energy, 1997, 36, 39–46 CAS.
- H. Zhang, G. A. Ye, H. F. Cong and T. Lan, Radiochim. Acta, 2012, 100, 813–820 CrossRef CAS.
- J. Bell and L. Toth, Radiochim. Acta, 1978, 25, 225–230 CrossRef CAS.
- Y. Wada, K. Wada, T. Goibuchi and H. Tomiyasu, J. Nucl. Sci. Technol., 1994, 31, 700–710 CrossRef CAS.
- T. Fukasawa and F. Kawamura, J. Nucl. Sci. Technol., 1991, 28, 27–32 CrossRef CAS.
- Y. Kitatsuji, T. Kimura and S. Kihara, J. Electroanal. Chem., 2010, 641, 83–89 CrossRef CAS.
- P. Masset, C. Apostolidis, R. Malmbeck and J. P. Glatz, J. Electroanal. Chem., 2007, 603, 166–174 CrossRef CAS.
- S. Y. Kim, T. Asakura, Y. Morita, G. Uchiyama and Y. Ikeda, J. Radioanal. Nucl. Chem., 2004, 262, 311–315 CrossRef CAS.
- N. E. Bibler, Nucl. Technol., 1977, 34, 412–415 CAS.
- G. Uchiyama, S. Fujine, S. Hotoku and M. Maeda, Nucl. Technol., 1993, 102, 341–352 CAS.
- G. Uchiyama, S. Hotoku, S. Fujine and M. Maeda, Nucl. Technol., 1998, 122, 222–227 CAS.
- A. Y. Zhang and Y. Liu, J. Radioanal. Nucl. Chem., 2000, 245, 357–361 CrossRef CAS.
- A. Y. Zhang, J. X. Hu and X. Y. Zhang, J. Radioanal. Nucl. Chem., 2002, 253, 107–113 CrossRef CAS.
- Y. X. Chen, H. B. Tang, J. P. Liu and H. He, J. Radioanal. Nucl. Chem., 2011, 289, 41–47 CrossRef CAS.
- Y. Ban, T. Asakura and Y. Morita, J. Radioanal. Nucl. Chem., 2009, 279, 423–429 CrossRef CAS.
- W. Q. Shi, H. B. Tang and Y. X. Ye, J. Nucl. Radiochem., 2002, 24, 134–137 CAS.
- V. S. Koltunov, K. M. Frolov and Y. V. Isaev, Radiochemistry, 2002, 44, 121–126 CrossRef CAS.
- V. S. Koltunov, S. M. Baranov and M. F. Tikhonov, Radiochemistry, 2002, 44, 248–252 CrossRef CAS.
- X. Y. Zhang, Z. L. Huang, S. T. Xiao and J. X. Hu, J. Nucl. Sci. Technol., 1998, 32, 433–438 CAS.
- X. Y. Zhang, G. A. Ye, S. T. Xiao and J. X. Hu, J. Nucl. Sci. Technol., 1997, 31, 193–198 CAS.
- V. Koltunov and S. Baranov, Inorg. Chim. Acta, 1987, 140, 31–34 CrossRef CAS.
- V. Koltunov, S. Baranov and T. Zharova, Radiochemistry, 1989, 31, 55–60 CAS.
- D. G. Yin, X. Y. Zhang and J. H. Hu, J. Nucl. Radiochem., 1997, 19, 23–28 CAS.
- Q. Y. Wu, J. H. Lan, C. Z. Wang, Z. P. Cheng, Z. F. Chai, J. K. Gibson and W. Q. Shi, Dalton Trans., 2016, 45, 3102–3110 RSC.
- J. A. Hlina, J. R. Pankhurst, N. Kaltsoyannis and P. L. Arnold, J. Am. Chem. Soc., 2016, 138, 3333–3345 CrossRef CAS PubMed.
- B. M. Gardner, G. Balazs, M. Scheer, F. Tuna, E. J. McInnes, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Nat. Chem., 2015, 7, 582–590 CrossRef CAS PubMed.
- C.-Z. Wang, W.-Q. Shi, J.-H. Lan, C.-L. Xiao, X.-K. Wang, Y.-L. Zhao, W.-Q. S. and Z.-F. Chai, Inorg. Chem., 2014, 53, 9607–9614 CrossRef PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS.
- J. Li, G. Schreckenbach and T. Ziegler, J. Am. Chem. Soc., 1995, 117, 486–494 CrossRef CAS.
- A. W. Ehlers and G. Frenking, J. Am. Chem. Soc., 1994, 116, 1514–1520 CrossRef CAS.
- B. Delley, M. Wrinn and H. P. Lüthi, J. Chem. Phys., 1994, 100, 5785–5791 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Gaussian, Inc., Wallingford, CT, USA, 2009 Search PubMed.
- X. Y. Cao and M. Dolg, J. Mol. Struct., 2004, 673, 203–209 CrossRef CAS.
- X. Y. Cao, M. Dolg and H. Stoll, J. Chem. Phys., 2003, 118, 487–496 CrossRef CAS.
- W. Küchle, M. Dolg, H. Stoll and H. Preuss, J. Chem. Phys., 1994, 100, 7535–7542 CrossRef.
- M. Dolg and X. Y. Cao, Chem. Rev., 2012, 112, 403–480 CrossRef CAS PubMed.
- N. Iché-Tarrat and C. J. Marsden, J. Phys. Chem. A, 2008, 112, 7632–7642 CrossRef PubMed.
- S. O. Odoh and G. Schreckenbach, J. Phys. Chem. A, 2010, 114, 1957–1963 CrossRef CAS PubMed.
- A. Klamt, J. Phys. Chem., 1995, 99, 2224–2235 CrossRef CAS.
- S. Sinnecker, A. Rajendran, A. Klamt, M. Diedenhofen and F. Neese, J. Phys. Chem. A, 2006, 110, 2235–2245 CrossRef CAS PubMed.
- R. Dennington, T. Keith and J. Millam, GaussView, version 5, Semichem Inc., Shawnee Mission, KS, 2009 Search PubMed.
- C. Gonzalez and H. B. Schlegel, J. Phys. Chem., 1990, 94, 5523–5527 CrossRef CAS.
- K. Fukui, Acc. Chem. Res., 1981, 14, 363–368 CrossRef CAS.
- A. Elkechai, Y. Mani, A. Boucekkine and M. Ephritikhine, Inorg. Chem., 2012, 51, 6943–6952 CrossRef CAS PubMed.
- K. Balasubramanian, W. J. Siekhaus and W. McLean, J. Chem. Phys., 2003, 119, 5889–5900 CrossRef CAS.
- C. F. Guerra, J. Snijders, G. Te Velde and E. Baerends, Theor. Chem. Acc., 1998, 99, 391–403 CAS.
- G. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. Fonseca Guerra, S. J. A. van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931–967 CrossRef CAS.
- A. Savin, R. Nesper, S. Wengert and T. F. Fässler, Angew. Chem., Int. Ed., 1997, 36, 1808–1832 CrossRef CAS.
- A. D. Becke and K. E. Edgecombe, J. Chem. Phys., 1990, 92, 5397–5403 CrossRef CAS.
- B. Silvi and A. Savin, Nature, 1994, 371, 683–686 CrossRef CAS.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- J. Cioslowski and S. T. Mixon, J. Am. Chem. Soc., 1991, 113, 4142–4145 CrossRef CAS.
- R. F. Bader, Acc. Chem. Res., 1985, 18, 9–15 CrossRef CAS.
- K. B. Wiberg, Tetrahedron, 1968, 24, 1083–1096 CrossRef CAS.
- I. Mayer, Int. J. Quantum Chem., 1984, 26, 151–154 CrossRef CAS.
- I. Mayer, Chem. Phys. Lett., 1983, 97, 270–274 CrossRef CAS.
- Q. Y. Wu, J. H. Lan, C. Z. Wang, Y. L. Zhao, Z. F. Chai and W. Q. Shi, J. Phys. Chem. A, 2015, 119, 922–930 CrossRef CAS PubMed.
- C. B. Braga, L. C. Ducati and R. Rittner, RSC Adv., 2015, 5, 18013–18024 RSC.
- D. Bhattacharjee, B. K. Mishra and R. C. Deka, RSC Adv., 2014, 4, 56571–56581 RSC.
- D. Cremer and E. Kraka, Angew. Chem., Int. Ed., 1984, 23, 627–628 CrossRef.
- D. Cremer and E. Kraka, Croat. Chem. Acta, 1984, 57, 1259–1281 Search PubMed.
- M. A. Mahi, S. M. Mekelleche, W. Benchouk, M. J. Aurell and L. R. Domingo, RSC Adv., 2016, 6, 15759–15769 RSC.
- S. Emamian, RSC Adv., 2015, 5, 72959–72970 RSC.
- L. R. Domingo, M. Ríos-Gutiérrez and J. A. Sáez, RSC Adv., 2015, 5, 37119–37129 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: The structures and related bond length of all species in the Pathways I, II and III (Fig. S1, S2 and S4 respectively), Np–O bond length of [NpVIO2(H2O)5]2+ and [NpVO2(H2O)5]2+ (Table S1), the QTAIM parameters and ELF pictures of Pathways II and III (Tables S2 and S4, Fig. S3 and S5) as well as the WBIs and Mayer bond orders (Table S3) are provided. See DOI: 10.1039/c6ra13339h |
| ‡ The first two authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |