
Monodisperse Ag–AgBr nanocrystals anchored on one-dimensional TiO2 nanotubes with efficient plasmon-assisted photocatalytic performance
Abstract
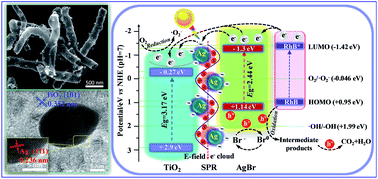
The broadening light absorption and efficient separation of photo-generated electron–hole pairs are crucial factors for the substantial applications of efficient artificial photocatalysts. Constructing novel nanoarchitectures with an appropriate interfacial junction and enhanced plasmonic fields exhibit promising behavior in photodegradation. Herein, we successfully synthesize a novel ternary Ag–AgBr/TiO2 plasmonic nanotube heterojunction photocatalyst by a facile deposition–precipitation strategy. Compared with the previously reported photocatalysts, the ternary Ag–AgBr/TiO2 plasmonic heterojunctions can thoroughly eliminate the organic pollutant RhB at an ultrafast degradation rate (the first-order reaction rate constant was calculated to be 0.1846 min−1). The main mechanism for the enhanced photocatalytic activity is that the unique tubular configuration not only provides a large specific surface area but also can contribute to the multiple diffractions and reflections of light and finally improve the light harvesting capability. Additionally, visible-light-driven AgBr anchored on the surface of TiO2 nanotubes, resulting in an intimate contact interface junction. Moreover, the stronger SPR effect of Ag nanoparticles significantly increases the production of photo-induced electron–hole pairs and effectively facilitates the plasmon-mediated interfacial electron transfer.
 Please wait while we load your content...
Please wait while we load your content...

