
Synthesis of azo linked polymers by a diazotization-coupling reaction and its application for CO2 capture
Abstract
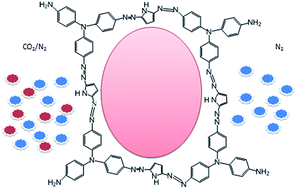
Azo bridged, heterocyclic, microporous polymers were synthesized by a metal catalyst-free direct one-step coupling reaction of a diazotized amine group with the five-membered ring. The two polymers were synthesized by incorporating pyrrole and imidazole groups. These polymers are amorphous in nature and exhibited surface areas between the range 100–300 m2 g−1. The CO2 adsorption capacity was investigated and it was 3.5 mmol g−1 at 298 K at 1 bar. Meanwhile, the CO2/N2 selectivity of the polymers was measured between 20 and 41 and the interactions of the polymers with gas molecules are discussed.
 Please wait while we load your content...
Please wait while we load your content...

