
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThiol-yne/thiol-epoxy hybrid crosslinked materials based on propargyl modified hyperbranched poly(ethyleneimine) and diglycidylether of bisphenol A resins
Cristina Aceboa,
Xavier Fernàndez-Francosb,
Xavier Ramis b and
Àngels Serra*ac
b and
Àngels Serra*ac
aDepartment of Analytical and Organic Chemistry, Universitat Rovira i Virgili, C/ Marcel·lí Domingo s/n, 43007, Tarragona, Spain. E-mail: angels.serra@urv.cat; Fax: +34 977558446; Tel: +34 077559558
bThermodynamics Laboratory, ETSEIB Universitat Politècnica de Catalunya, Av. Diagonal 647, 08028, Barcelona, Spain
cCentre Tecnològic de la Química de Catalunya, C/ Marcel·lí Domingo s/n, 43007, Tarragona, Spain
First published on 22nd June 2016
Abstract
A novel curing methodology based on the combination of thiol-yne and thiol-epoxy click reactions has been developed. The curing process consists of a first photoinitiated thiol-yne reaction, followed by a thermal thiol-epoxy process. As alkyne substrate a new propargyl terminated hyperbranched poly(ethyleneimine) (PEIyne) has been synthesized from the reaction of commercial poly(ethylenimine) (PEI) and glycidyl propargyl ether. The evolution of the curing of different mixtures of PEIyne and diglycidylether of bisphenol A (DGEBA) with the stoichiometric amount of tetrathiol (PETMP) has been monitored by DSC. The new hybrid materials have been characterized by thermomechanical analysis, thermogravimetry and by electronic microscopy inspection and compared with neat thiol-yne and thiol-epoxy materials. The Tgs of the complete cured materials increase with the proportion of epoxide in the formulation. The thiol-yne network improves the plasticity of the fracture of the materials.
1. Introduction
Cross-linked polymeric materials are extensively used in industrial applications.1 In the last few years more effort has been invested in obtaining materials for advanced technologies, and a growing interest in fully organic hybrid polymer networks has emerged. These hybrid polymeric materials can be obtained from the combination of two or more polymerization mechanisms of different reactive precursors. Usually, one of these mechanisms is initiated by photoirradiation and the other occurs by thermal treatment.2–4 Alternatively, two different photochemical processes can be combined5,6 or even two thermal polymerization steps.7 By combining polymerization mechanisms, it is possible to tailor the characteristics of the materials either by concurrent or sequential polymerization steps. The use of monomers or macromonomers with functionalities capable of participating in both polymerization mechanisms constitutes an advantage in the compatibilization of both polymeric structures that could have different polarity or miscibility.Thiol monomers can be used in a broad range of efficient synthetic processes with a great variety of reactive substrates like epoxides, acrylates, isocyanates, acetylenes and vinyl groups.8–10 Some of these reactions are radical-mediated, such as the photoinitiated thiol-ene and thiol-yne reactions or thermal nucleophilic processes, such as base-catalyzed thiol-epoxy, thiol-isocyanate and thiol-Michael additions. These reactions have been used, alone or combined, to prepare networked polymers in a fast and efficient manner due to the advantages associated to the click character of these reactions (fast, high conversion, solvent-free, oxygen-tolerant, etc.).11–13 In combined processes, multifunctional thiols, that can react in both stages of curing, play the role of coupling agents, connecting two types of polymeric structures, which hinders macrophase separation to occur.
The combination of thiol-ene and thiol-epoxy reactions to prepare crosslinked materials with tailored characteristics has been reported, but the networks formed are quite flexible and have a low crosslinking density, which leads to low glass transition temperatures.14–16
Recently, we reported the crosslinking of thiol-epoxy formulations containing allyl-terminated hyperbranched poly(ethyleneimine) (PEIene), used as a macromonomer, in thiol-ene/thiol-epoxy processes. The curing methodology consists in an initial photoinduced thiol-ene reaction followed by a thermal base catalyzed thiol-epoxy process.17 The advantage of using PEIene as a reactive component is to increase the functionality of the reactive mixture and enhance the thermomechanical characteristics of the intermediate material, since thiol-ene materials are generally highly flexible.18–20 Additionally, the use of hyperbranched polymers in epoxy thermosets modification has been reported to be advantageous to improve toughness and other mechanical characteristics.21,22
Thiol-yne reaction is a radical mediated click reaction which takes place in a two-step process: the addition of thiol to the carbon–carbon triple bond to yield an intermediate vinyl thioether, and a thiol-ene reaction with remaining thiol, yielding the 1,2-double-addition species. This reaction is well-documented in organic chemistry, but it has been seriously overlooked in the thermosets field. Recently, thiol-yne reaction has been used in the preparation of polyfunctional materials via sequential thiol-Michael/thiol-yne strategy. High refractive index optical networks suitable for diverse applications were obtained with high yield.23,24
To increase the functionality, in comparison with similar formulations based on thiol-ene process, and with the aim to explore the feasibility of novel combined thiol-epoxy and thiol-yne click processes, we have prepared propargyl terminated poly(ethyleneimine) (PEIyne) and added it in different proportions to thiol-epoxy formulations. The change from allyl to propargyl groups in the PEI structure should lead to an increase in the thermomechanical characteristics of the intermediate and final materials. The addition of two thiol groups to the propargyl moiety results in the double hydrothiolation of the triple bond and therefore the thiol-yne chemistry allows doubling the crosslinking density as compared with thiol-ene systems.25 We have selected the modification of NH groups of commercial poly(ethyeleneimine) to obtain propargyl terminated hyperbranched structures (PEIyne) as a more technological approach to the direct preparation of propargyl terminated hyperbranched polymers by thiol-halogen/thiol-yne condensation previously described.26
It is worth to note that the thiol-yne reaction, as it has been stated, has emerged as a useful tool for material synthesis for advanced applications, especially because of their good optical characteristics.27–31 However, to the best of our knowledge, there are no reported studies in the formation of hybrid networks from the combination of thiol-yne and thiol-epoxy reactions.
In the present study, mixtures in different proportions of diglycidyl ether of bisphenol A (DGEBA), PEIyne and tetrafunctional thiol (PETMP), catalyzed by a photoinitiator and a base, were photoirradiated and subsequently thermally cured. The evolution of the reactions was monitored by calorimetry in order to investigate if the two-stage curing process is sequential or both processes overlap. The materials prepared were characterized by thermomechanical analysis (DMTA), thermogravimetry (TGA) and their morphology visualized by electronic microscopy (SEM) and they were compared with neat thiol-yne and thiol-epoxy materials.
2. Experimental part
2.1 Materials
Poly(ethyleneimine) (PEI) Lupasol®FG (800 g mol−1) was kindly donated by BASF and used without further purification. From the molecular weight of the polymer and of the repeating unit, an average degree of polymerization of 18.6 was calculated. According to the data sheet, the relationship (NH2/NH/N) was (1/0.82/0.53) and thus by calculations the equivalent number of primary, secondary and tertiary amines resulted to be 10, 8.37 and 5.3 meq. g−1, respectively. Glycidyl propargyl ether (GPE), pentaerythritol tetrakis(3-mercaptopropionate) (PETMP), 1-methylimidazole (1-MI) and 2,2-dimethoxy-2-phenylacetophenone (DMPA) were all purchased from Sigma-Aldrich. Ethanol was purchased from Scharlab. Diglycidylether of bisphenol A (DGEBA), Araldite GY 240 (EEW = 182 g eq.−1) was provided by Huntsman.2.2 Synthesis of propargyl-terminated hyperbranched poly(ethyleneimine) (PEIyne) (Scheme 1)
In a 50 mL two neck round-bottomed flask provided with magnetic stirrer, addition funnel and Ar inlet, 1 g of PEI (1.25 mmol, 27.5 meq.) were dissolved in 10 mL of ethanol. Then, the stoichiometric quantity of glycidyl propargyl ether (3.08 g, 27.5 mmol) was added. The reaction mixture was kept at 50 °C for a day. The crude product (yellowish oil) was dried at 50 °C under vacuum during two days.1H NMR (CDCl3, δ in ppm) (see Fig. 1): 4.17 ppm (–O–CH2–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C, 4), 3.85 (–CH2–OH, 2), 3.48 ppm (–CH(OH)–CH2–O–, 3), 2.8–2.3 ppm (–CCH, 5; –CH2–N–, 1 and PEI nucleus).
C, 4), 3.85 (–CH2–OH, 2), 3.48 ppm (–CH(OH)–CH2–O–, 3), 2.8–2.3 ppm (–CCH, 5; –CH2–N–, 1 and PEI nucleus).
 | ||
| Fig. 1 1H NMR spectrum of propargyl-functionalized poly(ethyleneimine) PEIyne. | ||
13C NMR (CDCl3, δ in ppm): 80.3 (–![[C with combining low line]](https://www.rsc.org/images/entities/b_char_0043_0332.gif) CH), 78.9 ppm (–CH(OH)–H2–O–), 76.5 ppm (–O–H2–CH), 75.2 (–CH), 72.5 ppm (–H–OH–) and 70–50 ppm (–H2–N and PEI nucleus).
CH), 78.9 ppm (–CH(OH)–H2–O–), 76.5 ppm (–O–H2–CH), 75.2 (–CH), 72.5 ppm (–H–OH–) and 70–50 ppm (–H2–N and PEI nucleus).
Tg, determined by DSC = −40 °C.
Thermal stability, determined by TGA in N2, T5% = 321 °C, Tmax = 352 °C.
2.3 Preparation of the formulations and samples
PETMP–PEIyne in stoichiometric proportions were prepared and 1 phr of DMPA (1 part of photoinitiator for 100 parts of mixture) was added. The mixture was put in a bath at 70 °C until the solution became clear. PETMP–DGEBA stoichiometric mixture, containing 2 phr of 1-MI (parts of amine by 100 parts of DGEBA), was prepared by beating at room temperature. The curing formulations were prepared by beating at room temperature different amounts of the previously prepared PETMP–PEIyne and PETMP–DGEBA. Compositions of 20/80, 40/60, 60/40 and 80/20 w/w of PETMP–PEIyne/PETMP–DGEBA, neat PETMP–PEIyne and neat PETMP–DGEBA formulations were tested. The formulations were named as x thiol-yne, being x the proportion of PETMP-PEIyne mixture in the formulation.To prepare the stoichiometric formulations we have considered that the functionalities of the reactants are: 4 for PETMP, 2 for DGEBA and 22 for PEIyne. The composition of the mixtures is detailed in Table 1.
| Sample | PEIyne | DGEBA | PETMP | |||
|---|---|---|---|---|---|---|
| (mmol) | (g) | (mmol) | (g) | (mmol) | (g) | |
| 0 thiol-yne | — | — | 1.65 | 0.60 | 0.82 | 0.40 |
| 20 thiol-yne | 0.023 | 0.08 | 1.32 | 0.48 | 0.91 | 0.44 |
| 40 thiol-yne | 0.046 | 0.15 | 0.98 | 0.36 | 0.99 | 0.49 |
| 60 thiol-yne | 0.069 | 0.23 | 0.66 | 0.24 | 1.08 | 0.53 |
| 80 thiol-yne | 0.092 | 0.30 | 0.32 | 0.12 | 1.17 | 0.58 |
| 100 thiol-ene | 0.116 | 0.38 | — | — | 1.26 | 0.62 |
Rectangular samples of the crosslinked materials (40 mm × 10 mm × 1.5 mm) were obtained in a Teflon mold by irradiation under light-curing equipment (Dymax ECE 2000 UV light-curing flood lamp system) during 2 min (in intervals of 30 s with 5 min among them, 1 min for each face of the sample) (UV-intensity of 105 mW cm−2, 365 nm) and then thermally treated at 100 °C for 1 h and 120 °C for 30 min in an oven.
2.4 Characterization techniques
1H NMR and 13C NMR measurements were carried out in a Varian Gemini 400 spectrometer. CDCl3 was used as the solvent. For internal calibration the solvent signal corresponding to CDCl3 was used: δ (1H) = 7.26 ppm, δ (13C) = 77.16 ppm.Photocalorimetric experiments were performed in order to study the thiol-yne stage of the curing process. The samples were photocured at 30 °C using a Mettler DSC-821e calorimeter (Mettler-Toledo, Schwerzenbach, Switzerland) appropriately modified to permit irradiation with a Hamamatsu Lightningcure LC5 (HgeXe lamp) with two beams, one for the sample side and the other for the reference side. Samples weighing ca. 5 mg were cured in open aluminum pans in a nitrogen atmosphere. Two scans were performed on each sample in order to subtract the thermal effect of the UV irradiation from the photocuring experiment, each one consisting of 2 min of temperature conditioning, 6 min of irradiation and finally 2 more minutes without UV light. A light intensity of 30 mW cm−2 (calculated by irradiating graphite-filled pans on only the sample side) was employed.
The glass transition temperatures of the intermediate (after irradiation) and final material were determined by a dynamic scan from −100 °C to 250 °C, under a nitrogen atmosphere at 10 °C min−1 in a Mettler 822e device.
Dynamic postcuring experiments were carried out in a Mettler 822e calorimeter with a TSO801RO robotic arm (Mettler-Toledo, Schwerzenbach, Switzerland), from −100 °C to 250 °C, under a nitrogen atmosphere at 10 °C min−1. The degree of conversion, XUV, during the photocuring stage was calculated in the basis of the heat evolved during the postcuring as follows:
 | (1) |
A Jasco FTIR spectrometer equipment (resolution of 4 cm−1) with an attenuated-total-reflectance accessory with a diamond crystal (Golden Gate heated single-reflection diamond ATR, Specac-Teknokroma) was used to monitor the UV curing. All the measurements were performed at room temperature. An irradiation lamp (Hamamatsu Lightningcure LC5 (HgeXe lamp)) was used to induce the photopolymerization (light intensity of 30 mW cm−2). The samples were irradiated for 2 min and IR spectra were collected before and after irradiation. The UV process was followed by the thiol band at 2570 cm−1 and propargyl band at 2109 cm−1.
Thermogravimetric analyses (TGA) were carried out in a nitrogen atmosphere with a Mettler TGA/SDTA 851e thermobalance. Samples with an approximate mass of 8 mg were degraded between 30 and 800 °C at a heating rate of 10 °C min−1 in N2 (100 cm3 min−1 measured in normal conditions).
Dynamic mechanical thermal analyses (DMTA) were carried out with a TA Instruments DMA Q800 device. The samples were prepared as described before. Single cantilever bending at 1 Hz and amplitude 10 μm was performed at 3 °C min−1 from 50 °C below Tg to 50 °C after Tg for each sample.
The cryofracture of the specimens was metalized with gold and observed with a scanning electron microscopy (SEM) Jeol JSM 6400 with a resolution of 3.5 nm.
3. Results and discussion
3.1 Synthesis and characterization of the propargyl-terminated hyperbranched poly(ethyleneimine) (PEIyne)
In a previous work,32 we synthesized a propargyl-terminated poly(ethylenimine) of higher molecular weight (Lupasol® G100, 5000 g mol−1) by reaction of PEI with propargyl acrylate. In that case, we proved that the functionality in propargyl units achieved was only 58.6 in average, which means that NH2 and NH react only with one acrylate group. Since in the present study we aim to increase the functionality of the components of the mixture to achieve higher Tgs, we modified PEI following a similar synthetic procedure that the one used in the preparation of allyl terminated PEI,17 that is, through the reaction of PEI with propargyl glycidyl ether as shown in Scheme 1. Using this methodology, NH2 reacts twice with glycidyl groups and NH once, reaching a practically complete modification. In the present study, we started from PEI with lower molecular weight because of viscosity issues, since the first reaction in the curing process is a photoinitiated thiol-yne at room temperature without any solvent. | ||
| Scheme 1 Synthetic route to propargyl terminated hyperbranched poly(ethyleneimine). | ||
The characterization of the PEIyne synthesized was performed by FTIR and NMR spectroscopy. By FTIR the propargyl group produced a characteristic band at 2110 cm−1 and the presence of hydroxyl groups was put in evidence by a broad adsorption centered at 3300 cm−1.
Fig. 1 shows the 1H NMR spectrum of PEIyne in CDCl3. As we can see, methylene and methine protons of the new unit appear as separate signals at 4.17, 3.85 and 3.48 ppm, but acetylenic protons (5) appear overlapped with the signals corresponding to methylene protons linked to N of the PEI structure.
The degree of modification was calculated by 1H NMR spectroscopy, similarly as described in a previous paper17 but taking into account that the signals corresponding to the propargyl proton overlapped with the signals of the core and should be subtracted. The modification degree calculated by this methodology was approximately 100%, taking into account the range of error attributable to the determination procedure. According to that, the number of propargyl groups per PEIyne molecule is 22 in average.
3.2 Study of the photopolymerization process
Thiol-yne reaction takes place by a radical-mediated step-growth mechanism initiated by photoirradiation. The reaction proceeds under mild conditions, rapidly and achieve a high yield, without side-reactions, similarly to what happens in thiol-ene reaction, but with a double addition of thiol groups to triple bond.11 Type II photoinitiators, like benzophenone derivatives, can be used to initiate this process. The initiator absorbs UV light and goes to the triplet state, where it abstracts hydrogen from the thiols forming thiyl radicals. Both, thiol-ene and thiol-yne reactions proceed by the addition of a thiyl radical to the substrate. As illustrated in Scheme 2, after the first addition, a carbon-centered radical is produced that subsequently abstracts a hydrogen from another thiol, generating a vinyl sulfide moiety and regenerating a thiyl radical. The vinyl sulfide is capable of undergoing further reaction through the addition of a second thiyl radical to the vinyl sulfide. Each yne moiety reacts with two thiol groups to form an 1,2-dithioether structure.23,25 The reaction proceeds at high rates under ambient humidity and atmospheric oxygen conditions to high conversion, thereby providing an extremely efficient methodology for fabricating high-performance cross-linked polymer networks and films in a facile and convenient fashion. | ||
| Scheme 2 Reaction mechanism proposed for the addition of thiols to alkynes. | ||
From the kinetics point of view, the rate of alkyne disappearance is equal to the rate of thiol loss and vinyl creation. The formed vinyl sulfides are consumed immediately upon creation and therefore alkyne acts as a difunctional in thiol-yne step-growth polymerizations.25 Bowman's group demonstrated that thiol-yne materials have a higher crosslink density, glass transition temperature and modulus as compared to an analogous thiol-ene network.25
The monitoring of thiol-yne reaction was performed by FTIR and photocalorimetry. A mixture in stoichiometric proportions of PEIyne and PETMP and 1 phr of DMPA was irradiated in the UV chamber during 120 s (in intervals of 30 s with 5 min between them, 60 s irradiation on each face of the sample). The material obtained was studied by FTIR. Fig. 2 shows the FTIR spectra before and after irradiation.
 | ||
| Fig. 2 FTIR spectra of the 100 thiol-yne formulation, before and after UV irradiation. | ||
The typical signals of thiol groups (at 2576 cm−1) and acetylene units (at 2110 cm−1), which are magnified, disappear completely, indicating that the fully reaction has been achieved. No absorptions of vinyl sulfide groups can be detected. The UV irradiation of this mixture in the photocalorimeter at 30 °C produces an enthalpy of about 122 kJ eq.−1 of triple bond, just twice the enthalpy released in the thiol-ene polymerization system previously studied by us.17 The Tgs after scanning in the photo-DSC and after irradiation in the chamber were determined and the value of 25 °C was obtained for both materials (see Table 2). The multifunctionality of the PEIyne structure allows to reach higher Tgs than those described for networks prepared from simple tetrafunctional thiols and alkyne formulations.33 Once the most adequate conditions to perform thiol-yne reaction were achieved, the curing of the complex mixtures was undertook.
| Sample | DSC data after irradiation in photo-DSC | DSC data after irradiation in the chamber | Final material | ||||
|---|---|---|---|---|---|---|---|
| ΔHa (kJ per CCeq.) |
Xb (%) | Tgc (°C) | ΔHd (kJ per ee) | Xe (%) | Tgf (°C) | Tgg (°C) | |
| a Heat released per yne equivalent during irradiation in the photo-DSC during 6 min at 30 °C.b Epoxy group conversion after 2 min of irradiation in the photo-DSC determined by eqn (1).c Glass transition of the material after 2 min of irradiation in photo-DSC determined in a second dynamic scan.d Heat released by epoxy equivalent after irradiation during 2 min in the irradiation chamber, determined in a dynamic scan at 10 °C min−1.e Epoxy group conversion after irradiation in the chamber determined from DSC data and eqn (1).f Glass transition of the intermediate material after irradiation during 2 min in the chamber determined by DSC.g Glass transition determined in a second scan in the DSC at 10 °C min−1, of the final material after irradiation in the chamber. | |||||||
| 0 thiol-yne | — | — | 125 | — | — | 56 | |
| 20 thiol-yne | 122 | 3 | −27 | 120 | 4 | −27 | 54 |
| 40 thiol-yne | 121 | 5 | −26 | 118 | 6 | −23 | 35 |
| 60 thiol-yne | 121 | 17 | −6 | 102 | 18 | −4 | 29 |
| 80 thiol-yne | 124 | 33 | −2 | 80 | 36 | 2 | 28 |
| 100 thiol-yne | 122 | — | 25 | — | — | 25 | 25 |
3.3 Study of combined photo-thermal processes
To establish a new sequential photo/thermal dual curing process, it is desired that both reactions occur independently in a controlled manner, with a stable intermediate material.3,16 Up to now, no description on the use of thiol-yne/thiol-epoxy sequences in thermoset preparation has been reported, but the procedure must be parallel to thiol-ene/thiol-epoxy two step curing.The main issue in the combination of both click reactions in a two-step curing process is that the initial thiol-yne reaction, which is photoinitiated, releases heat. Thus, some thiol-epoxy reaction could occur during this photoirradiation, because of the presence of tertiary amines in the PEI structure and the added 1-MI which can catalyze the thermal process, as depicted in Scheme 3. In the previous study with PEIene it was put in evidence that 1-MI must be added to the formulation to reach the complete thiol-epoxy reaction.
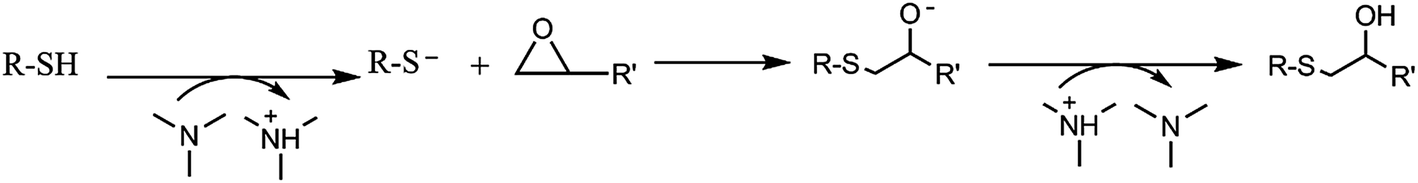 | ||
| Scheme 3 Mechanism of epoxy-thiol reaction catalyzed by tertiary amines. | ||
In the thiol-ene/thiol-epoxy process with PEIene, both reactions partially overlap and therefore the intermediate material is not only consequence of the thiol-ene, but also thiol-epoxy contributes to the formation of the network.17 Therefore, in the present case, it is also required the investigation of the possible overlapping. To do that, different PEIyne/DGEBA mixtures in different proportions with the stoichiometric proportion of PETMP were studied by photo-DSC. The enthalpy released was measured and the enthalpy per alkyne equivalent calculated. The values obtained are collected in Table 2. As we can see, the values of enthalpy are similar, which could indicate that the thiol-yne process reached the completeness, even when a high proportion of epoxide is present in the formulation. By thermal dynamic DSC scans performed from each irradiated mixture, the heat evolved in the thiol-epoxy thermal process was evaluated. From these values we were able to calculate the conversion of epoxide groups during irradiation by using eqn (1) and taking into account the enthalpy of the neat epoxy-thiol formulation (0 thiol-yne) and its proportion in the combined curing process. As we can see in Table 2, the conversion in epoxide during the UV stage increases with the proportion of thiol-yne reaction and it is even higher than the one calculated in PEIene/DGEBA/PETMP formulations.17 This fact seems to support that the heat released during the UV-process, initiates the thermal reaction. In the previous study, we could determine that formulations with 20 and 40 of thiol-ene reaction behave in the photocalorimeter as dual curing systems, in a sequential and well controlled manner. This does not happen in the present thiol-yne/thiol-epoxy formulations.
FTIR studies gave us no more insight in the chemistry of this process, since the mixtures were rather complex and the most typical bands weak.
The preparation of the samples to be further characterized was performed by irradiating in the UV chamber in molds. After irradiation, the samples were investigated by dynamic DSC to determine the extent in which the epoxy conversion has occurred in these conditions by measuring the remaining enthalpy. The values of enthalpy determined are collected in Table 2, together with the conversion of epoxide calculated as explained before. As we can see, the extent of epoxy-thiol reaction is similar in the UV chamber to the one determined in the photocalorimeter, which is different from that we obtained in PEIene/DGEBA formulations.
The Tgs of the intermediate materials, after irradiation in photoDSC and UV chamber were determined and they are collected in Table 2. As it can be seen, they increase with the amount of PEIyne in the formulation, as the result of the higher crosslinking by thiol-yne reaction on the hyperbranched structure of the multifunctional PEIyne and are similar in both cases. However, not only the thiol-yne network is the responsible of the Tg achieved, but the competitive thiol-epoxy reaction also contributes to the increase of this value. If we look at the Tg of the completely cured material we can appreciate a tendency of increasing on increasing the proportion of epoxide in the formulation until reaching similar values of 54–56 °C for both 20 thiol-yne and 0 thiol-yne samples. The Tgs of the final materials prepared with the contribution of thiol-yne reaction are slightly higher than the ones obtained by thiol-ene, when the proportion of radical mediated process is predominant.17 It should be taken into account that thiol-yne formulations are richer in PETMP than thiol-ene, which is the more flexible structure participating in the global network and therefore the increase in Tg is limited.
3.4 Characterization of the photo-thermal cured materials
The thermal stability of the materials obtained after irradiation in the UV chamber and thermally cured at 100 °C for 1 h and 120 °C for 30 min in an oven was studied by thermogravimetry. Fig. 3 shows the weight-loss curves and their derivatives.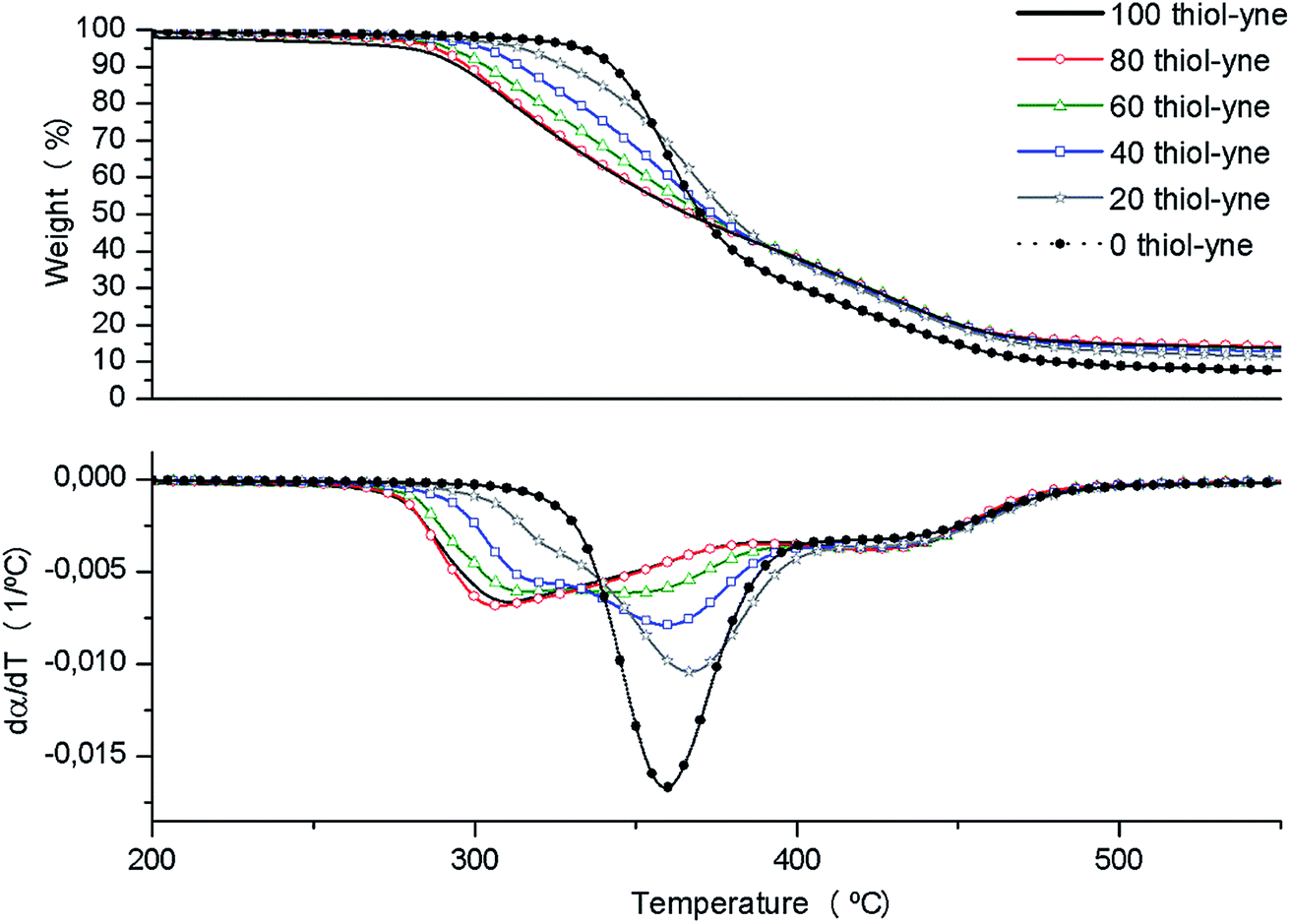 | ||
| Fig. 3 Thermogravimetric curves at 10 °C min−1 in N2 atmosphere of cured materials. | ||
From the thermogravimetric curves we can see that the addition of PEIyne to the formulations leads to more degradable materials, since the neat DGEBA/PETMP (0 thiol-yne) thermoset begins to degrade at the highest temperature with a more simple shape of the derivative curve. When PEIyne was in the formulation the initial decomposition temperature diminishes, the range of decomposition temperatures broadens, and the mechanism of degradation becomes more complex. This behavior changes accordingly to the composition of the materials. This behavior was the one expected, since we have previously observed the lower thermal stability of thermosets modified with PEI derivatives, because of the breakage of C–N bonds.17,34
The temperature-dependent viscoelastic properties of the materials prepared were characterized by DMTA. The mechanical spectra, including plots of E′, E′′ and tan![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) δ against temperature for all materials are shown in Fig. 4.
δ against temperature for all materials are shown in Fig. 4.
 | ||
| Fig. 4 tanδ evolution, loss and storage moduli against the temperature for the materials obtained. | ||
The addition of PEIyne to the formulation leads to a reduction of the temperature of the tanδ maximum and to the broadening of the curve indicating a higher flexibility and a less homogeneous character of the network, according to the contribution of the hyperbranched poly(ethyleneimine) to the network structure.
From the loss modulus plots, the peaks corresponding to the Tgs can be observed. The evolution of this parameter clearly reflects the composition of the formulation. The higher the proportion of PEIyne the lower the Tg of the materials because of the flexibility introduced, although the reduction of this value in the 20 thiol-yne material in reference to the neat 0 thiol-yne is scarce, such as obtained from DSC studies.
Storage modulus plots show a broad relaxation for the materials with thiol-yne proportions higher than 40%. Usually, E′ values reach a plateau at a temperature about 50 °C higher than the main relaxation temperature. The values of moduli at this plateau decrease with decreasing cross-link density of the networks, consistent with the prediction of the thermodynamic equation of state for ideal rubber elasticity.35 However, in the present case the complexity of the network with thiol-yne, thiol-epoxy and poly(ethyleneimine) structures does not lead to any regular variation in the storage moduli in the rubbery region. However, neat thiol-epoxy (0 thiol-yne) materials, with a high molecular weight between crosslinks show the lowest value of relaxed modulus in comparison to the hybrid formulations. Accordingly, 100 thiol-yne, with the lowest molecular weight between crosslinks leads to the highest value.
The morphologies of the cryofractured materials were visualized by scanning electron microscopy (SEM). Fig. 5 shows the most representative micrographs of the samples.
 | ||
| Fig. 5 SEM micrographs of the cryofractured surfaces of some of the cured materials prepared. | ||
As we can observe, whereas the neat thiol-epoxy material presents a quite fragile fracture with a little evidence of deformation, the samples containing PEIyne show a highly plastic fracture, which must enhance the toughness of the materials. The SEM micrograph at higher magnifications of the sample 20 thiol-yne in Fig. 6 shows a nanograined morphology produced by the hybrid character of the network structure. The other hybrid samples also showed some type of nanophase separation.
 | ||
| Fig. 6 SEM micrograph showing the morphology of the 20 thiol-yne sample. | ||
4. Conclusions
Propargyl units were introduced at the chain ends of hyperbranched poly(ethyleneimine) by reaction of this polymer with glycidyl propargyl ether, achieving a nearly complete modification, as evaluated by 1H NMR spectroscopy. This propargyl terminated HBP, PEIyne, was used as a macromonomer in a thiol-yne photopolymerization reaction.Different formulations of PEIyne and DGEBA in different proportions with the stoichiometric amount of PETMP were firstly photoirradiated and then thermally cured to obtain hybrid organic materials. The curing process was a combination of a photoinitiated thiol-yne and a thermally induced thiol-epoxy click reaction, catalyzed by a photoinitiator (DMPA) and 1-MI as a base, respectively.
The study of this curing process allowed us to confirm that in the photoirradiation step, in which thiol-yne reaction occurred, thiol-epoxy reaction competed, due to the heat evolved in the thiol-yne coupling and the presence of tertiary amines in the formulation.
The double addition of thiol groups to the alkyne moieties allowed reaching higher glass transition temperatures in the materials in comparison to similar formulations based on thiol-ene reactions, when thiol-yne process was predominant, accordingly to their higher crosslinking densities.
The Tgs of the cured materials increased on decreasing the proportion of PEIyne in the formulation, because of the more rigid structure of DGEBA.
The thermal stability is decreased on increasing the proportion of PEIyne in the formulation due to the presence of PEI structures.
The addition of PEIyne to the materials increased the plastic character of the fracture, which must enhance the toughness of these materials. The composition of the hybrid materials led to the formation of nanostructures, which could be observed by SEM microscopy.
Acknowledgements
The authors would like to thank MINECO (MAT2014-53706-C03-01, MAT2014-53706-C03-02) and Generalitat de Catalunya (2014-SGR-67) for giving financial support. BASF and Huntsman are acknowledged for kindly providing Lupasol samples and Araldite GY 240, respectively.References
- M. Chanda and S. K. Roy, Industrial Polymers, Specialty Polymers and Their Applications, CRC Press, Boca Raton, USA, 2007 Search PubMed.
- M. Adachi, H. Okamura and M. Shirai, Chem. Lett., 2013, 42, 1056–1058 CrossRef CAS.
- G. González, X. Fernández-Francos, À. Serra, M. Sangermano and X. Ramis, Polym. Chem., 2015, 6, 6987–6997 RSC.
- C.-H. Park, S.-W. Lee, J.-W. Park and H.-J. Kim, React. Funct. Polym., 2013, 73, 641–646 CrossRef CAS.
- B. de Ruiter, A. El-ghayoury, H. Hofmeier, U. S. Schubert and M. Manea, Prog. Org. Coat., 2006, 55, 154–159 CrossRef CAS.
- R. Acosta Ortiz, A. E. Garcia Valdéz, L. Berlanga Duarte, R. Guerrero Santos, L. R. O. Flores and M. D. Soucek, Macromol. Chem. Phys., 2008, 209, 2157–2168 CrossRef.
- X. Fernández-Francos, A.-O. Konuray, A. Belmonte, S. De la Flor, À. Serra and X. Ramis, Polym. Chem., 2016, 7, 2280–2290 RSC.
- J. Xu and C. Boyer, Macromolecules, 2015, 48, 520–529 CrossRef CAS.
- G.-Z. Li, R. K. Randev, A. H. Soeriyadi, G. Rees, C. Boyer, Z. Tong, T. P. Davis, C. R. Becer and D. M. Haddleton, Polym. Chem., 2010, 1, 1196–1204 RSC.
- A. H. Soeriyadi, G.-Z. Li, S. Slavin, M. W. Jones, C. M. Amos, C. R. Becer, M. R. Whittaker, D. M. Haddleton, C. Boyer and T. P. Davis, Polym. Chem., 2011, 2, 815–822 RSC.
- Thiol-X Chemistries in Polymer and Materials Science, ed. A. B. Lowe and C.N. Bowman, RSC Publishing, Croydon, UK, 2013 Search PubMed.
- C. E. Hoyle, A. B. Lowe and C. N. Bowman, Chem. Soc. Rev., 2010, 39, 1355–1387 RSC.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS PubMed.
- F. Saharil, F. Forsberg, Y. Liu, P. Bettotti, N. Kumar, F. Niklaus, T. Haraldsson, W. van der Wijngaart and K. B. Gylfason, J. Micromech. Microeng., 2013, 23, 025021 CrossRef.
- J. A. Carioscia, J. W. Stansbury and C. N. Bowman, Polymer, 2007, 48, 1526–1532 CrossRef CAS PubMed.
- D. Guzmán, X. Ramis, X. Fernández-Francos and A. Serra, RSC Adv., 2015, 5, 101623–101633 RSC.
- C. Acebo, X. Fernández-Francos, X. Ramis and A. Serra, React. Funct. Polym., 2016, 99, 17–25 CrossRef CAS.
- T. Yoshimura, T. Shimasaki, N. Teramoto and M. Shibata, Eur. Polym. J., 2015, 67, 397–408 CrossRef CAS.
- B. G. Rutherglen, R. A. McBath, Y. L. Huang and D. A. Shipp, Macromolecules, 2010, 43, 10297–10303 CrossRef CAS.
- D. A. Shipp, C. W. McQuinn, B. G. Rutherglen and R. A. McBath, Chem. Commun., 2009, 6415–6417 RSC.
- L. Boogh, B. Pettersson and J. A. E. Månson, Polymer, 1999, 40, 2249–2261 CrossRef CAS.
- M. Flores, X. Fernández-Francos, F. Ferrando, X. Ramis and A. Serra, Polymer, 2012, 53, 5232–5241 CrossRef CAS.
- A. B. Lowe, C. E. Hoyle and C. N. Bowman, J. Mater. Chem., 2010, 20, 4745–4750 RSC.
- J. W. Chan, C. E. Hoyle and A. B. Lowe, J. Am. Chem. Soc., 2009, 131, 5751–5753 CrossRef CAS PubMed.
- D. B. Fairbanks, T. F. Scott, C. J. Kloxin, K. S. Anseth and C. N. Bowman, Macromolecules, 2009, 42, 211–217 CrossRef PubMed.
- Y. Zheng, S. Li, Z. Weng and C. Gao, Chem. Soc. Rev., 2015, 44, 4091–4130 RSC.
- A. B. Lowe, Polymer, 2014, 55, 5517–5549 CrossRef CAS.
- J. W. Chan, H. Zhou, C. E. Hoyle and A. B. Lowe, Chem. Mater., 2009, 21, 1579–1585 CrossRef CAS.
- Q. Wei, R. Pötzsch, X. Liu, H. Komber, A. Kiriy, B. Voit, P.-A. Will, S. Lenk and S. Reineke, Adv. Funct. Mater., 2016, 26, 2545–2553 CrossRef CAS.
- R. Hoogenboom, Angew. Chem., Int. Ed., 2010, 49, 3415–3417 CrossRef CAS PubMed.
- R. Pötzsch, H. Komber, B. C. Stahl, C. J. Hawker and B. Voit, Macromol. Rapid Commun., 2013, 34, 1772–1778 CrossRef PubMed.
- C. Acebo, A. Lederer, D. Appelhans, X. Ramis and A. Serra, send to revision.
- J. W. Chan, J. Shin, C. E. Hoyle, C. N. Bowman and A. B. Lowe, Macromolecules, 2010, 43, 4937–4942 CrossRef CAS.
- X. Fernández-Francos, D. Santiago, F. Ferrando, X. Ramis, J. M. Salla, A. Serra and M. Sangermano, J. Polym. Sci., Part B: Polym. Phys., 2012, 50, 1489–1503 CrossRef.
- L. H. Sperling, Introduction to Physical Polymer Science, John Wiley & Sons, New York, USA, 2005 Search PubMed.
| This journal is © The Royal Society of Chemistry 2016 |