
Symmetric and unsymmetric thienyl-substituted fluorenone dyes: static excimer-induced emission enhancement†
Fan Xua,
Mao-Sen Yuan*ab,
Wenji Wanga,
Xianchao Dua,
Hui Wanga,
Na Lia,
Ruijin Yua,
Zhenting Du a and
Jinyi Wang*a
a and
Jinyi Wang*a
aCollege of Science, Northwest A&F University, Yangling 712100, P. R. China. E-mail: yuanms@nwsuaf.edu.cn; jywang@nwsuaf.edu.cn; Fax: +86 29 87082520; Tel: +86 29 87082520
bState Key Laboratory of Crystal Materials, Shandong University, Jinan 250100, PR China
First published on 9th August 2016
Abstract
In this work, we designed and synthesized four symmetric and asymmetric thienyl-substituted fluorenone compounds, which all exhibited typical AIE properties. Besides a high solid-state fluorescence quantum yield, the enhanced luminescence of their solid-state powders showed a 170 nm red-shift (from 380 to 550 nm) in comparison with the luminescence of their dilute tetrahydrofuran solutions. The photophysical properties and single-crystal structure, combined with the theoretical calculation, revealed that the bathochromic luminescence is due to the formation of static excimers.
Introduction
Research on organic luminescent materials has been paid increasing attention because of their vast potential in the fields of organic electronics,1–3 optoelectronics4,5 and biology6,7 etc. Any breakthrough in luminescent materials undoubtedly promotes the generation of high-tech products and benefits society.8–10 The design and synthesis of new luminescent materials is of continuing importance. One thing to note is that most of the organic light-emitting materials need to be used in the solid state or aggregation state in practical application, for example, organic light-emitting devices (OLEDs),11–14 organic laser crystals,15 light detectors16–18 and communication equipment,19 and so on. However, for conventional light-emitting systems, aggregation will cause partial or complete quenching of fluorescence.20 The phenomenon of aggregation-caused quenching (ACQ) has been documented for more than half a century since Förster's discovery of the concentration quenching effect in 1954.21 From the viewpoint of real-world applications, however, it creates numerous problems.20,22In 2001, the anti-ACQ phenomenon of aggregation-induced emission (AIE), a non-emissive compound in organic solution that exhibits an obviously enhanced fluorescence in its aggregated state, was first reported by Tang and co-workers.23 The AIE phenomenon has been a hot research topic in recent years. This discovery was undoubtedly an important milestone in the history of solid-state light-emitting materials. In recent years, many AIE compounds have been found successively, such as the hexaphenylsilole (HPS),24 tetraphenylethene (TPE),25 tetraphenylpyrazine,25 cyano-stilbene26 and difluoroboron avobenzone27 compounds. Whereas a wealth of information has been collected, much remains to be understood and explored. Simultaneously, compared with the discovery of new AIE compounds, deciphering the AIE process is even more important. As long as the mechanism is clearly known, then we can effectively design and synthesize AIE luminescent materials. Up to now, the working principles of AIE have been explored and understood to some extent, and a number of possible mechanisms have been proposed, including restriction of intramolecular rotation (RIR)28–32 and intramolecular vibrations,33 conformational planarization,34,35 relatively loose molecular packing,36,37 J-aggregate formation38–40 and special excimer formation.41–44 Among them, the RIR mechanism has been experimentally verified repeatedly.45 Actually, the luminescence of most AIE compounds follows the RIR mechanism. But not all AIE situations can be explained by RIR. Although some different mechanisms have been proposed, the studies so far have been woefully insufficient. Therefore, it is necessary to design experiments and strategies to conduct further research on these mechanisms.
Our works intend to further expand the family of AIE compounds, together with research on the photophysical properties and the luminescent mechanism of the new AIE compounds. In previous works, Tao and our group reported a class of phenyl-substituted fluorenone compounds with symmetric molecular configuration, which exhibited typical AIE properties.41,42,46 Herein, in order to further understand the character and internal mechanism of AIE, we designed and synthesized several symmetric and asymmetric thienyl-substituted fluorenone compounds:
5-(9-oxo-9H-fluoren-2-yl)thiophene-2-carbaldehyde (CTPF),
2-(5-acetylthiophen-2-yl)-9H-fluoren-9-one (ATPF),
5′-(9-oxo-9H-fluorene-2,7-diyl)bis(thiophene-2-carbaldehyde) (BTPF), and
1,1′-((9-oxo-9H-fluorene-2,7-diyl)bis(thiophene-5,2-diyl))bis(ethan-1-one) (ETPF).
Thiophene-based optoelectronic materials have been extensively investigated because of their good thermal and chemical stability, as well as their high luminous efficiency. On the other hand, the influence of molecular symmetry on AIE properties is one of the factors we intend to explore. Unlike traditional AIE compounds, these thienyl-substituted fluorenone compounds show a ∼170 nm bathochromic enhanced fluorescence in comparison with their dilute THF solution. The photophysical properties and single-crystal structure, combined with the theoretical calculations, revealed that the bathochromic luminescence is due to the formation of static excimer.
Experimental
Materials and characterization
Solvents for reactions and spectral measurements were dried and distilled before use. The reagents used for reactions were purchased from J&K Scientific Ltd. 1H NMR spectra were recorded at 25 °C on Bruker Avance 500 MHz spectrometer using CDCl3 and CF3COOD (TFA) as solvent. 13C NMR spectra were recorded at 25 °C on Bruker Avance 125 MHz spectrometer using CDCl3 and CF3COOD as solvent. Element analyses (C, H) were performed using a PE 2400 autoanalyser. Mass spectrometry analyses were performed by a Bruker Biflex III matrix assisted laser desorption/ionization time of flight (MALDI-TOF) mass spectrometer.Synthesis of CTPF
The synthesis of the fluorescent CTPF dye was as follows: 2,7-dibromo-9H-fluoren-9-one (0.300 g, 0.888 mmol) and (5-formylthiophen-2-yl)boronic acid (0.415 g, 2.663 mmol) were dissolved in the mixture of toluene (20 mL), ethanol (5 mL) and 2 M potassium hydroxide aqueous solution (1 mL). The mixture was stirred at room temperature for 0.5 h under N2 gas followed by the addition of Pd(PPh3)4 (5 mg, 4.33 × 10−3 mmol) and then heated to 80 °C for 20 h. After completion, the mixture was poured into water and extracted three times with dichloromethane. The organic layer was dried over anhydrous sodium sulfate. After removing the solvent under vacuum, the residue was chromatographed on a silica gel column with CH2Cl2 as eluent to give CTPF (0.185 g, 72% yield). Melting point (mp): 235.5–237.1 °C. IR (KBr, cm−1): 3467, 3322, 3088, 1707, 1638, 1597, 1433, 1281, 804, 738. 1H NMR (500 MHz, CDCl3, ppm): δ 7.40 (t, J = 7.0 Hz, 1H), 7.52 (d, J = 3.9 Hz, 1H), 7.67–7.55 (m, 3H), 7.76 (d, J = 7.3 Hz, 1H), 7.89–7.79 (m, 2H), 8.02 (s, 1H), 9.96 (s, 1H). 13C NMR (CDCl3, 125 MHz, ppm): δ 190.6, 182.7, 143.7, 142.8, 137.3, 135.1, 134.4, 132.4, 129.7, 124.7, 124.6, 122.0, 121.1, 120.8. TOF-MS-EI: m/z 291.0 [M]+. Elemental anal. calcd for C18H10O2S: C, 74.46 and H, 3.47. Found: C, 74.65 and H, 3.28.Synthesis of ATPF
The synthesis of the fluorescent ATPF dye was as follows: 2,7-dibromo-9H-fluoren-9-one (0.300 g, 0.888 mmol) and (5-acetylthiophen-2-yl)boronic acid (0.453 g, 2.663 mmol) were dissolved in the mixture of toluene (20 mL), ethanol (5 mL) and 2 M potassium hydroxide aqueous solution (1 mL). The mixture was stirred at room temperature for 0.5 h under N2 gas followed by the addition of Pd(PPh3)4 (5 mg, 4.33 × 10−3 mmol) and then heated to 80 °C for 20 h. After completion, the mixture was poured into water and extracted three times with dichloromethane. The organic layer was dried over anhydrous sodium sulfate. After removing the solvent under vacuum, the residue was chromatographed on a silica gel column with methyl alcohol–CH2Cl2 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :200 by volume) as eluent to give ATPF (0.188 g, 70% yield). Melting point (mp): 215.6–217.4 °C. IR (KBr, cm−1): 3389, 3066, 2923, 2853, 1715, 1659, 1604, 1437, 1221, 815, 735, 676. 1H NMR (500 MHz, CDCl3, ppm): δ 2.63 (s, 3H), 7.39 (dd, J = 7.3, 6.3 Hz, 1H), 7.44 (d, J = 3.9 Hz, 1H), 7.66–7.53 (m, 3H), 7.74 (dd, J = 8.0, 5.7 Hz, 2H), 7.83 (dd, J = 7.8, 1.7 Hz, 1H), 8.00 (d, J = 1.3 Hz, 1H). 13C NMR (CDCl3, 125 MHz, ppm): δ 194.3, 190.5, 144.5, 143.5, 141.0, 139.7, 135.1, 134.37, 133.4, 132.3, 129.5, 126.4, 124.6, 124.5, 121.8, 121.0, 120.7, 26.6. TOF-MS-EI: m/z 305.1 [M]+. Elemental anal. calcd for C19H12O2S: C, 74.98 and H, 3.97. Found: C, 74.75 and H, 4.12.
:200 by volume) as eluent to give ATPF (0.188 g, 70% yield). Melting point (mp): 215.6–217.4 °C. IR (KBr, cm−1): 3389, 3066, 2923, 2853, 1715, 1659, 1604, 1437, 1221, 815, 735, 676. 1H NMR (500 MHz, CDCl3, ppm): δ 2.63 (s, 3H), 7.39 (dd, J = 7.3, 6.3 Hz, 1H), 7.44 (d, J = 3.9 Hz, 1H), 7.66–7.53 (m, 3H), 7.74 (dd, J = 8.0, 5.7 Hz, 2H), 7.83 (dd, J = 7.8, 1.7 Hz, 1H), 8.00 (d, J = 1.3 Hz, 1H). 13C NMR (CDCl3, 125 MHz, ppm): δ 194.3, 190.5, 144.5, 143.5, 141.0, 139.7, 135.1, 134.37, 133.4, 132.3, 129.5, 126.4, 124.6, 124.5, 121.8, 121.0, 120.7, 26.6. TOF-MS-EI: m/z 305.1 [M]+. Elemental anal. calcd for C19H12O2S: C, 74.98 and H, 3.97. Found: C, 74.75 and H, 4.12.
Synthesis of BTPF
The synthesis of the fluorescent BTPF dye was as follows: 2,7-dibromo-9H-fluoren-9-one (0.300 g, 0.888 mmol) and (5-formylthiophen-2-yl)boronic acid (0.415 g, 2.663 mmol) were dissolved in the mixture of toluene (20 mL), ethanol (5 mL) and 2 M potassium acetate aqueous solution (1 mL). The mixture was stirred at room temperature for 0.5 h under N2 gas followed by the addition of Pd(PPh3)4 (5 mg, 4.33 × 10−3 mmol) and then heated to 80 °C for 20 h. The red solid gradually precipitated from the reaction mixture. Subsequently, the reaction mixture was subjected to suction filtration with sand core funnel and after filtration to give BTPF of red solid powder. The red solid was rinsed three times with dichloromethane. The yield of compound BTPF: 0.277 g, 78%. Melting point (mp): >300 °C. IR (KBr, cm−1): 3409, 3101, 2837, 1715, 1658, 1604, 1563, 1418, 1226, 1052, 807, 774, 672. 1H NMR (500 MHz, TFA, ppm): δ 7.65 (dd, J = 19.9, 5.5 Hz, 2H), 7.94 (d, J = 7.7 Hz, 1H), 8.10–7.98 (m, 2H), 9.91 (s, 1H). 13C NMR (TFA, 125 MHz, ppm): δ 187.1, 180.9, 155.9, 145.1, 141.9, 140.5, 134.5, 134.1, 125.5, 122.6, 121.7. TOF-MS-EI: m/z 401.0 [M]+. Elemental anal. calcd for C23H12O3S2: C, 68.98 and H, 3.02. Found: C, 68.84 and H, 2.97.Synthesis of ETPF
The synthesis of the fluorescent ETPF dye was as follows: 2,7-dibromo-9H-fluoren-9-one (0.300 g, 0.888 mmol) and (5-acetylthiophen-2-yl)boronic acid (0.453 g, 2.663 mmol) were dissolved in the mixture of toluene (20 mL), ethanol (5 mL) and 2 M potassium acetate aqueous solution (1 mL). The mixture was stirred at room temperature for 0.5 h under N2 gas followed by the addition of Pd(PPh3)4 (5 mg, 4.33 × 10−3 mmol) and then heated to 80 °C for 20 h. The red solid gradually precipitated from the reaction mixture. Subsequently, the reaction mixture was subjected to suction filtration with sand core funnel and after filtration to give ETPF of red solid powder. The red solid was rinsed three times with dichloromethane. The yield of compound ETPF: 0.284 g, 75%. Melting point (mp): 298.3–300.5 °C. IR (KBr, cm−1): 3413, 3284, 3082, 1715, 1651, 1433, 1275, 1026, 798, 747, 598. 1H NMR (500 MHz, TFA, ppm): δ 2.68 (s, 3H), 7.40 (d, J = 3.2 Hz, 1H), 7.47 (d, J = 7.4 Hz, 1H), 7.73 (d, J = 8.9 Hz, 2H), 7.85 (d, J = 3.2 Hz, 1H). 13C NMR (TFA, 125 MHz, ppm): δ 196.9, 196.4, 153.6, 144.4, 141.1, 136.9, 134.2, 133.4, 125.1, 122.0, 121.5, 24.2. TOF-MS-EI: m/z 429.1 [M]+. Elemental anal. calcd for C25H16O3S2: C, 70.07 and H, 3.76. Found: C, 69.78 and H, 3.62.Photophysical properties measurement
UV-vis absorption spectra for the solutions and the films were recorded with a Shimadzu UV-2550 spectrophotometer. Photoluminescence (PL) spectra were recorded using a Shimadzu RF-5301PC spectrofluorimeter. The fluorescence quantum yield (Φ) in solution was determined using rhodamine B in ethanol as a reference according to a previously reported method.47 Quantum yield of the solid-state powder was determined with a PTI C-701 calibrated integrating sphere system.48 Steady-state fluorescence spectra and decay curves were obtained using an Edinburgh FLS920 fluorescence spectrometer equipped with a time-correlated single photon counting (TCSPC) card. Reconvolution fits of the decay profiles were performed with F900 analysis software to obtain the lifetime values.Single crystal X-ray diffraction
The single crystal of compound ATPF were obtained by the slow diffusion of their respective THF/cyclohexane solutions for several days at room temperature. Since the crystal is stable under ambient condition, the data collection was done without any inert gas protection at room temperature on a Bruker SMART APEX-II CCD area detector using graphite-monochromated Mo Ka radiation (λ = 0.71073 Å). Data reduction and integration, together with global unit cell refinements were done by the INTEGRATE program of the APEX2 software. Semi-empirical absorption corrections were applied using the SCALE program for area detector. The structure was solved by direct methods and refined by the full matrix least-squares methods on F2 using SHELX.49Theoretical calculation
In order to understand the spectral behavior and elucidate the influence of the configurational change on the spectra, the orbital energy of the single-molecule and dimer of compound ATPF were respectively calculated by using the Gaussian 09 program at the B3LYP Time-Dependent Density Functional Theory (TD-DFT). The geometries of the ATPF single-molecule and the ATPF dimer were obtained from its determined X-ray single crystal structure.Results and discussion
Synthesis of CTPF, ATPF, BTPF and ETPF
The molecular structures of the compounds CTPF, ATPF, BTPF and ETPF are outlined in Scheme 1. They were readily synthesized through a conventional Suzuki reaction by reacting 2,7-dibromo-9H-fluoren-9-one with the corresponding thienyl boric acid. All the new compounds were characterized with 1H NMR, 13C NMR, elemental analysis and MALDI/TOF mass spectroscopy. Compound ATPF has been verified by the X-ray single-crystal structure.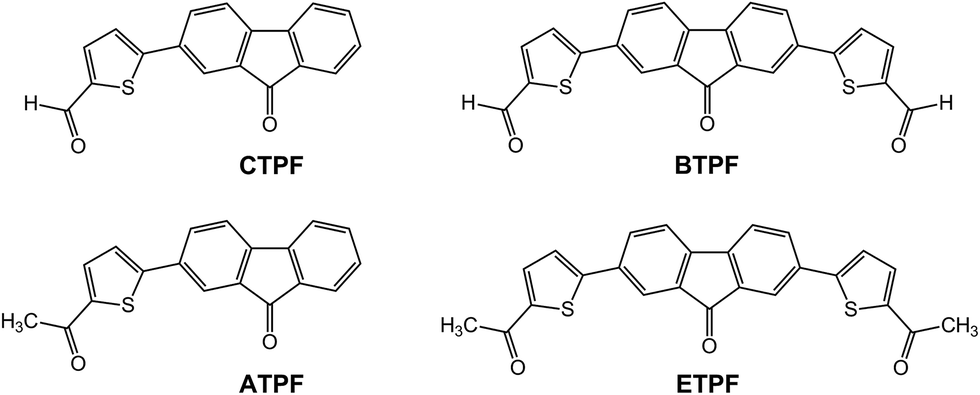 | ||
| Scheme 1 Molecular structures of compounds CTPF, ATPF, BTPF and ETPF. | ||
AIE and photophysical properties
The synthesized compounds of CTPF, ATPF, BTPF and ETPF have the typical characteristic that they contain ketone groups. According to the research of Zojer,50 in these ketone-containing fluorenone compounds, S0 → S1 corresponds to an optically forbidden transition and possesses an n–π* character, the energy gap of which is lower than that of the charge-transfer π–π* state. This case will result in weak single-molecular fluorescence. As shown in Fig. 1a and b, the fluorescence of these four fluorenone compounds in dilute THF solution is really weak under 365 nm UV light (Table 1, fluorescence quantum yield Φ ranging from 3% to 8%), and they give very weak PL spectra with an emission maximum (λem) of 380 nm (Fig. 1d). However, the solid powder of all the symmetric and asymmetric compounds showed bright luminescence with the fluorescence quantum yields Φ ranging from 38% to 78% (Fig. 1b and Table 1), which indicated that they have the typical characteristics of the AIE compounds.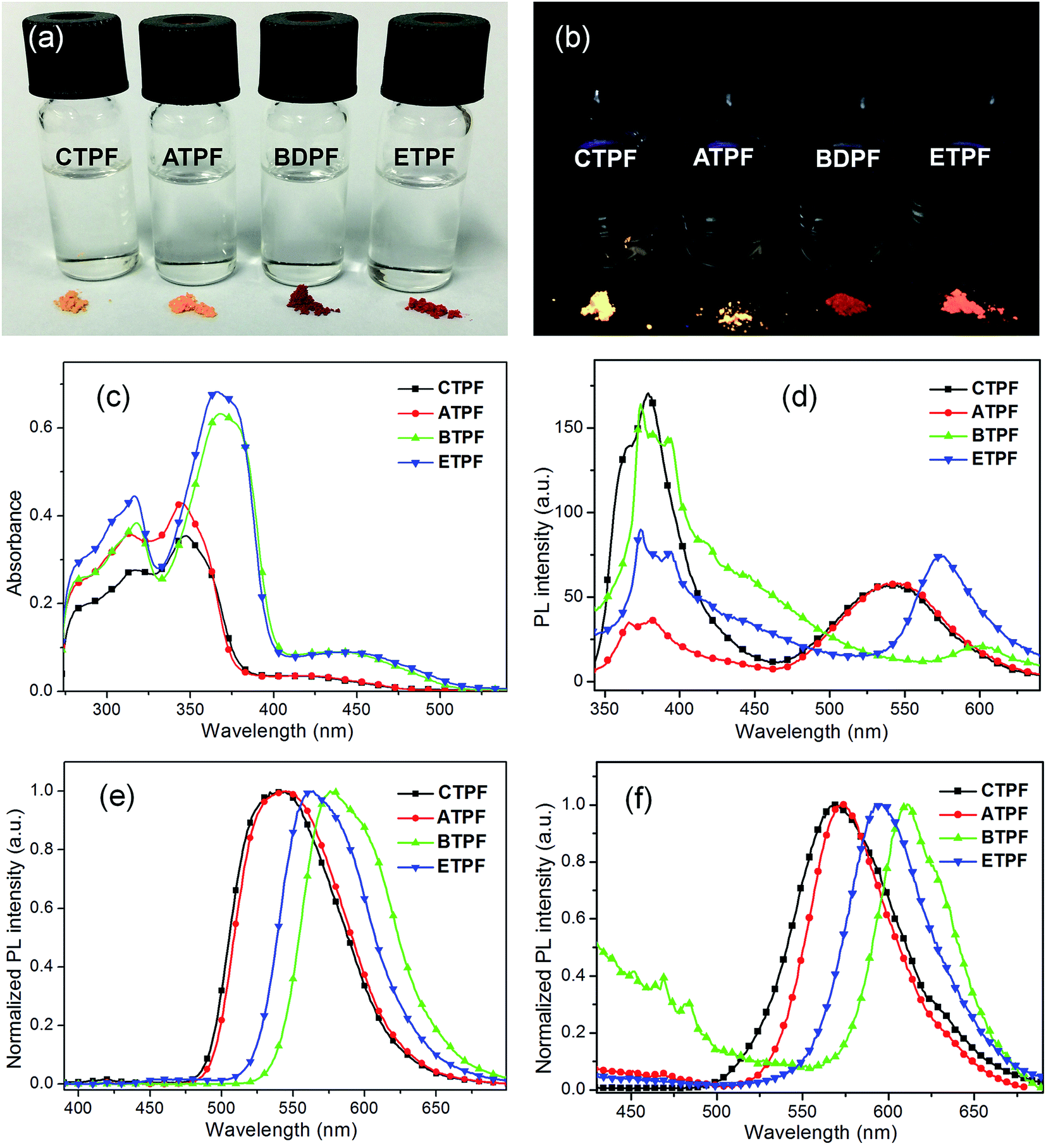 | ||
| Fig. 1 (a) Photos of the THF solutions (10 μM) and solid powders of compounds CTPF, ATPF, BTPF and ETPF under natural light and (b) 365 nm UV light. (c) UV-visible absorption spectra and (d) PL spectra of the THF solutions (10 μM) of compounds CTPF, ATPF, BTPF and ETPF. (e) Normalized PL spectra of the THF solutions (100 μM) of compounds CTPF, ATPF, BTPF and ETPF. (f) Normalized PL spectra of the solid powders of compounds CTPF, ATPF, BTPF and ETPF. | ||
| Solution in THFa | Solution in THF/watera (80% water) | Crystalline powder | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| λabs (nm) | λem (nm) | Φb | τc (ns) | λabs (nm) | λem (nm) | Φ | λem (nm) | Φd | τ (ns) | |
| a With c = 1.0 × 10−5 mol L−1.b Fluorescence quantum yield was determined using rhodamine B in ethanol as a standard.c Fluorescence lifetime.d Fluorescence quantum yield in the solid state was obtained using a calibrated integrating sphere. | ||||||||||
| CTPF | 317, 347, 433 | 379, 542 | 0.03 | 0.24 | 298, 351, 464 | 547 | 0.42 | 570 | 0.78 | 3.53 |
| ATPF | 313, 345, 435 | 366, 382, 544 | 0.04 | 0.21 | 296, 352, 469 | 548 | 0.44 | 573 | 0.71 | 4.02 |
| BDPF | 318, 368, 447 | 374, 393, 599 | 0.07 | 0.41 | 298, 354, 471 | 570 | 0.41 | 611 | 0.38 | 3.35 |
| EDPF | 316, 366, 450 | 374, 394, 574 | 0.08 | 0.39 | 298, 357, 470 | 556 | 0.40 | 595 | 0.52 | 3.91 |
In addition, we also noted that the PL spectra of their 10 μM THF solutions exhibited two primary emission bands with peaks at ∼380 and 550 nm respectively (Fig. 1d). The 380 nm emission showed an obvious vibronic structure, and the 550 nm emission did not. In order to further figure out the origination of the two different PL peaks, the PL spectra of their concentrated THF solutions have also been determined. As we all know, when the concentration of the solution increases, the distance between the molecules in solution will become less, and the interaction between molecules will strengthen. Fig. 1e shows the PL spectra of the 100 μM THF solution of the four compounds. We can clearly see that, with the concentration increasing, the PL spectral shapes of the four compounds changed greatly compared to those of their 10 μM THF solutions. The emission peak at ∼380 nm almost disappeared and the emission peak at ∼550 nm markedly increased. The solid powder of the four compounds exhibited almost the same emissive character as their 100 μM THF solution, but red-shifted to a certain extent (Fig. 1f). Their respective emission peaks are consistent with the fluorescence images of their solid powder in Fig. 1b.
We also measured the mixed system of THF and water at 10 μM to further understand the spectral characteristics of the four thienyl-substituted fluorenone compounds. The water fractions are respectively 30%, 40%, 50%, 60%, 70%, 80% and 90%. As water is a poor solvent for these four compounds, the high ratio of water will lead to the gathering of molecules. As shown in Fig. 2a, with the increasing water content, the 300 nm absorption intensity of the CTPF compound decreased and an obvious level-off tail formed in the long-wavelength region of the absorption spectra. Synchronously, we observed that the violet PL peak at 380 nm progressively vanished and the blue PL peak at 550 nm was gradually boosted in the PL spectra (Fig. 2b). The above results indicate that the PL peak at 380 nm originates from single molecules, while the PL peak at 550 nm originates from the aggregation.
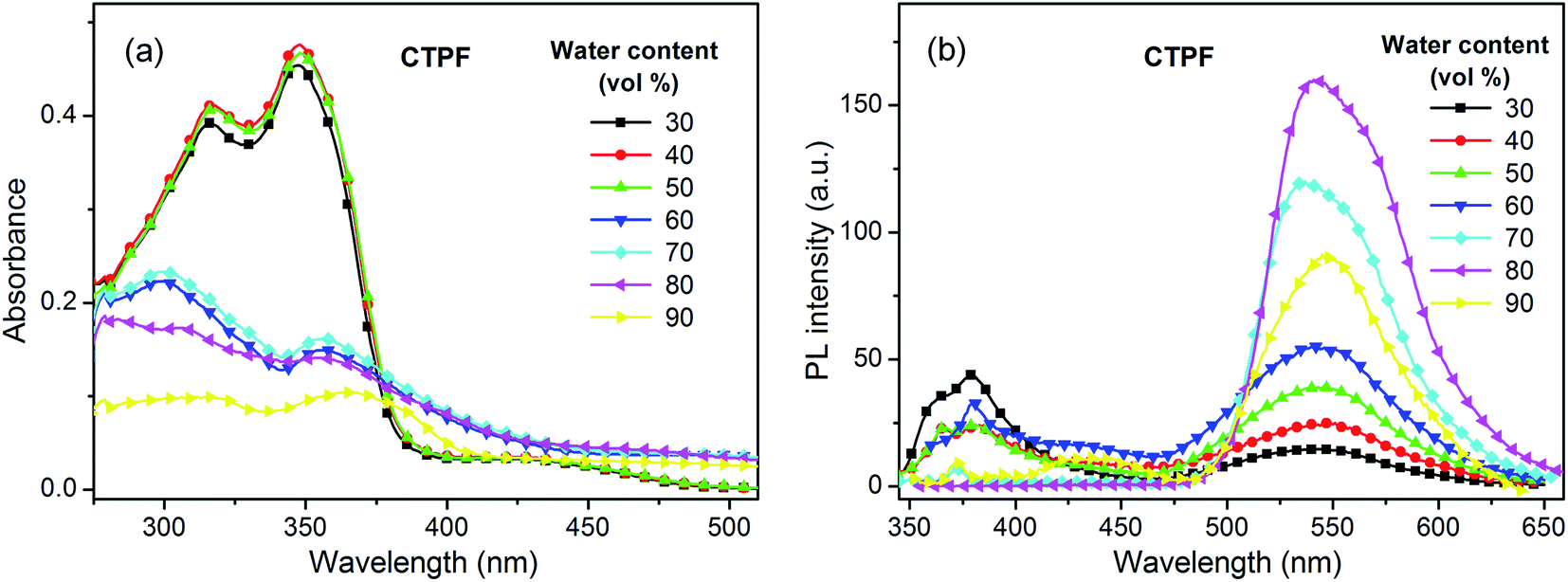 | ||
| Fig. 2 (a) UV-visible absorption spectra changes of CTPF depending on the water fractions in THF. (b) PL spectra changes of CTPF depending on the water fractions in THF. Compounds ATPF, BTPF and ETPF exhibited similar character to CTPF. | ||
In addition, we noted that the four thienyl-substituted fluorenone dyes displayed different spectral behavior from the phenyl-substituted fluorenone compounds reported by us. The four thienyl-substitutions exhibited characteristic double-peaks emission peaked at ∼380 and 550 nm respectively in their dilute THF solutions (Fig. 1d). However, the reported phenyl-substitutions showed only single-peak emission in their dilute THF solutions.41,46 The result indicates that the thienyl-substitutions are more likely to aggregation than the phenyl-substitutions. Moreover, the aggregation state of the four thienyl-substituted fluorenone dyes cannot be easily dismissed when diluting, and will eventually reach a balance between the single molecules and the aggregations in solutions.
On the other hand, the 170 nm bathochromic phenomena that appeared in both cases are different from the spectral behaviours of common AIE dyes, whose enhanced luminescence is caused by the restricted intramolecular rotation of peripheral aromatic rings or the effects of intramolecular planarization in their aggregated states. In those AIE systems, the peak positions of their PL maxima do not change much, in either the single-molecule state or the aggregate state. In addition, we measured the fluorescence lifetime of these four compounds in solid-state and dilute THF solution (10 μM) and found, as shown in Fig. 3, that the fluorescence lifetime of these four compounds in dilute THF solution is much shorter than the solid powder, which indicates that these four compounds experienced different decay routes from the excited state to the ground state in the two different states.
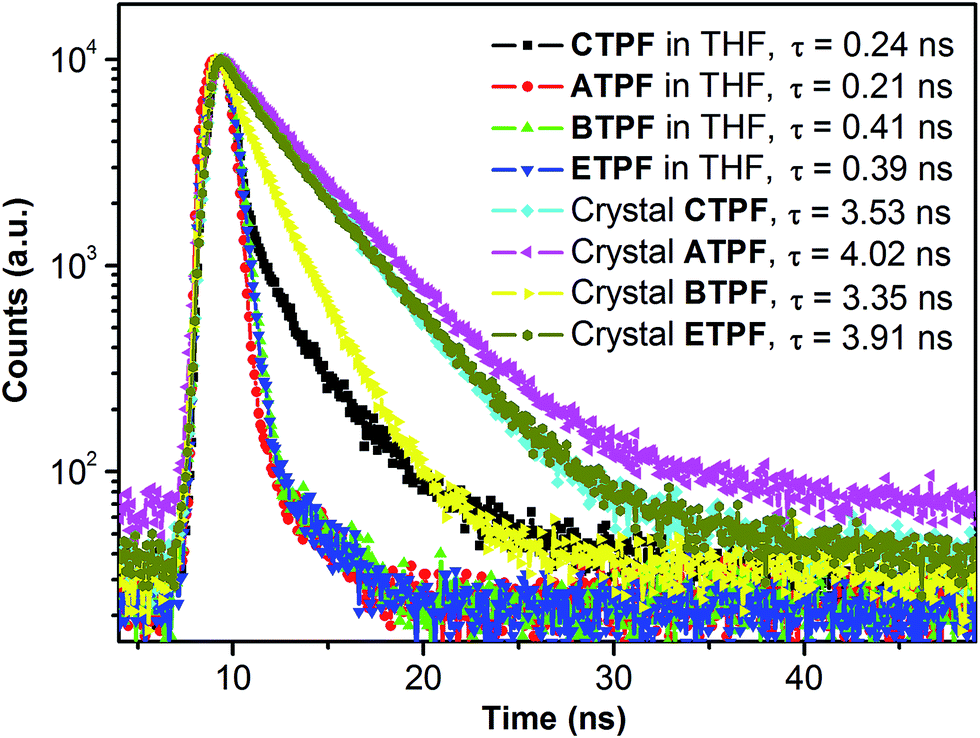 | ||
| Fig. 3 Fluorescence lifetime profiles of compounds CTPF, ATPF, BTPF and ETPF in THF (10 μM) and in the solid state respectively. | ||
X-ray crystallography and AIE mechanism
Determination of the X-ray single-crystal structure is the most effective and convincing tool for acquiring ground-state molecular structures and exploring the AIE mechanism. Single crystals of ATPF were obtained by slow diffusion of their THF/cyclohexane (1:1 v/v) solutions for several days at room temperature.51 The molecular packing of ATPF is shown in Fig. 4. The ATPF molecule exhibited a highly planar geometry, and the dihedral angle between the thiophene ring and the fluorenone ring is only 5.1(2)°. These molecules were packed into molecular columns through intermolecular π–π stacking (Fig. 4a). Surprisingly, the contact distances between the two up-down adjacent molecules are not the same, being respectively 3.39 Å and 3.65 Å. We think that the up-down molecular pairs with a closer contact distance form dimers. And the dimers are further stacked into molecular columns based on weak π–π interaction with 3.65 Å contact distance. Upon being excited, these dimers are turned into excimers. Generally, such π–π stacking structure or excimer formation will reduce the molecular luminous efficiency due to the non-radiation transition. However, the ATPF solid powder exhibited a high fluorescence quantum yield (71%). Usually, two molecules of conventional excimer combine together after being excited, and then need to be properly rearranged or suitably restructured, and structural adjustment is also needed in the decay process. Such a process will consume a large part of the excitation energy, which will lead to non-radiation transition and fluorescence quenching. In the ATPF crystal structure, π–π stacked molecular pairs overlap with the electronegative five-membered ring (which involves the carbonyl group of the five-membered ring) that faces the electropositive thiophene ring (Fig. 4c), and directly form a static excimer in the ground. When the excimers are excited, they do not undergo any energy-consuming configurational rearrangement, resulting in fluorescence enhancement. Moreover, weak C–H⋯O hydrogen bonds also exist between two adjacent molecular columns (Fig. 4d). These intermolecular hydrogen bonds further stabilize the molecular configuration and impede the molecular structural adjustment after being excited.
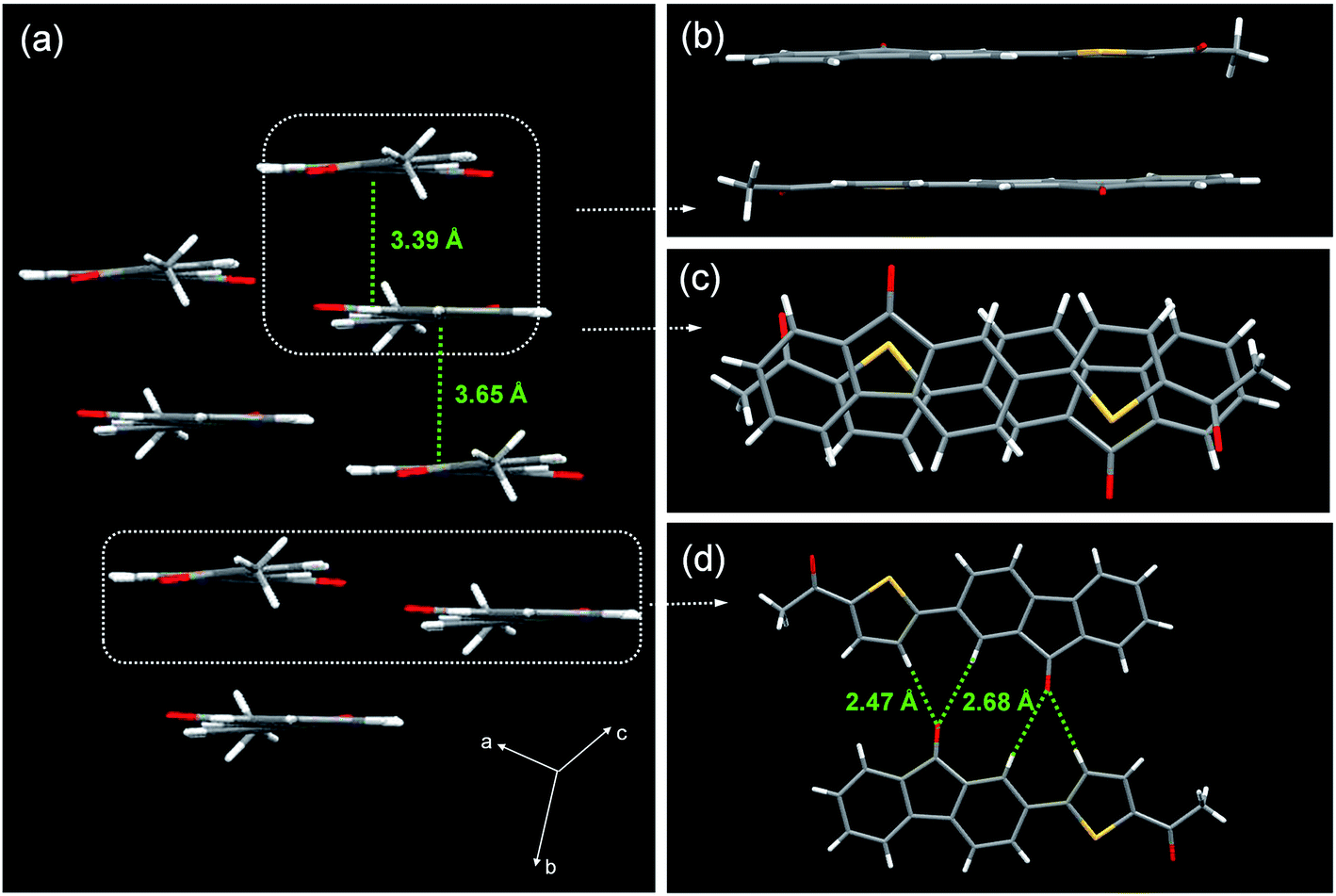 | ||
| Fig. 4 X-ray crystallographic packing of compound ATPF. (a) Side view of ATPF crystal packing. (b) Top view and illustration of the hydrogen bonding in ATPF. Side (c) and top (d) view and illustration of the π–π stacking in ATPF. | ||
Theoretical studies
To better understand the spectral behaviour and inspect whether the great bathochromic solid-state emission of the four fluorenone compounds is derived from the dimers, we respectively calculated the molecular orbital energies of the ATPF single-molecule and the ATPF dimer (the geometry was directly obtained from its X-ray single crystal structure) using the Gaussian 09 program and time-dependent density functional theory (TD-DFT). As shown in Fig. 5, the energy gap of the ATPF single-molecule between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) is 3.45 eV (359 nm). When the two ATPF molecules form a dimer via π–π stacking interactions, the energy gap of the ATPF dimer between the HOMO and LUMO is reduced by 0.63 eV (81 nm) compared to that of ATPF single-molecule. Upon the ATPF dimer being excited, charge transfer occurs from one molecule to the other molecule through the intermolecular π⋯π channel. The results accord well with their spectral characteristics, and both the absorption and emission of solid powder ATPF display an obvious red shift compared with those of its dilute THF solution (Fig. 2c).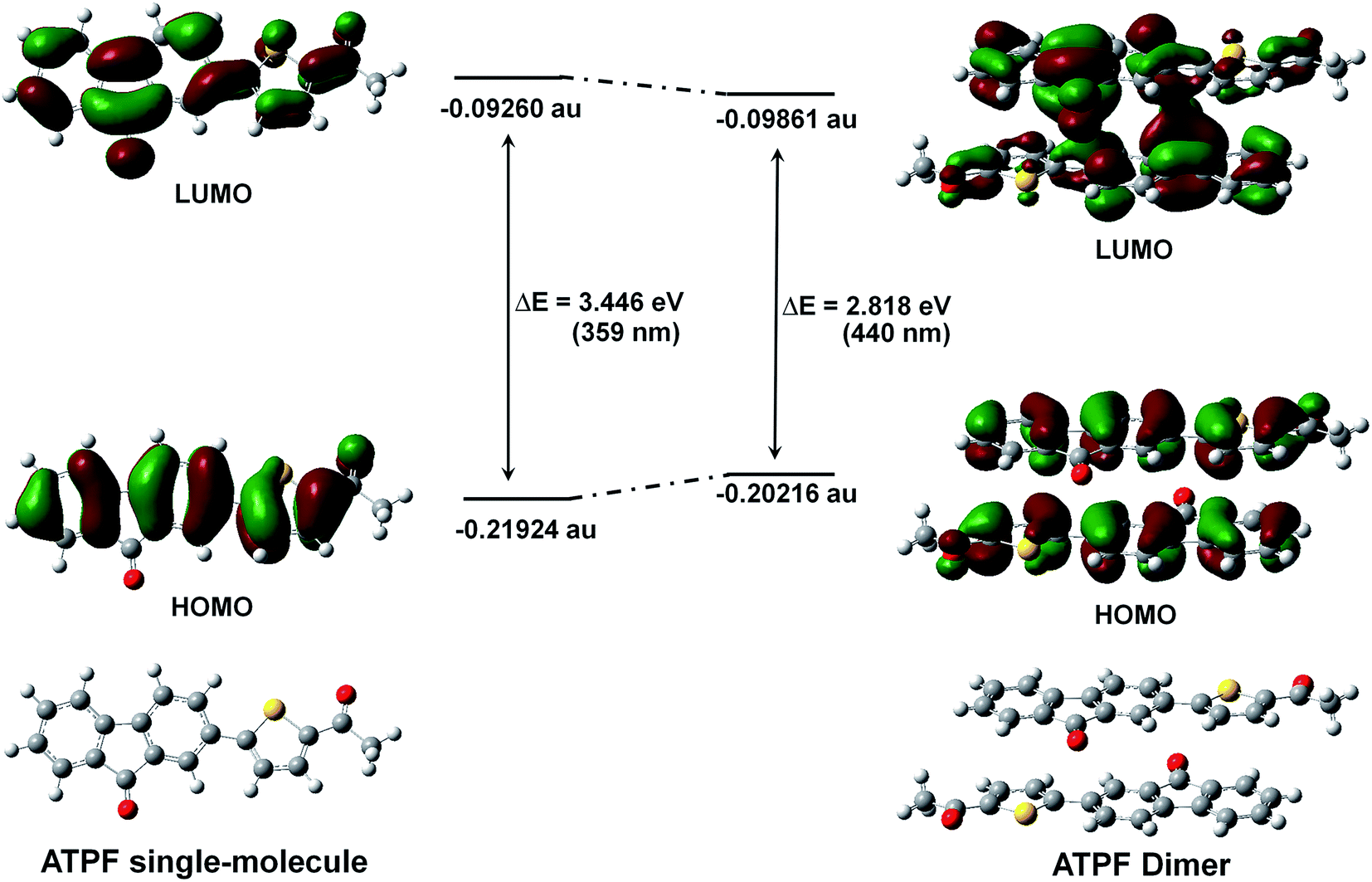 | ||
| Fig. 5 The LUMOs, HOMOs and molecular geometries of ATPF single-molecule and ATPF dimer (the geometry was obtained from its X-ray single crystal structure directly) with their relative energy according to TD-DFT calculation. | ||
Conclusions
In this study, we designed and synthesized four symmetric and asymmetric thienyl-substituted fluorenone compounds, namely CTPF, ATPF, BTPF and ETPF. All four compounds exhibit typical AIE characteristics with high solid-state fluorescence quantum yields. Not only is the process of molecular aggregation accompanied by a gradual increase in the fluorescence intensity, but also the peak in the aggregate-state has a 170 nm red-shift with respect to single molecules. We sought the reason by measuring the X-ray single crystal structure of ATPF and conducting theoretical calculations. In the crystal packing, we found that every two up-down adjacent molecules are bound together even in the ground state by π–π stacking interactions to form a static excimer (dimer). We believe that the bathochromic fluorescent enhancement is derived from the static excimers. Furthermore, by calculating the molecular orbital energies of the ATPF single-molecule and the ATPF dimer, the computed results are in good agreement with the spectral characteristics of both absorption and emission.Acknowledgements
This study was supported by the National Natural Science Foundation of China (21202132, 21175107 and 21375106), the Fund of Youth Science and Technology Stars by Shaanxi Province (2015KJXX-15), the Ministry of Education of the People's Republic of China (NCET-08-602 0464), China Postdoctoral Science Foundation (2016M592837).Notes and references
- F. Cicoira and C. Santato, Adv. Funct. Mater., 2007, 17, 3421–3434 CrossRef CAS.
- Z. Zhao, J. W. Y. Lam and B. Z. Tang, J. Mater. Chem., 2012, 22, 23726–23740 RSC.
- S. J. Ananthakrishnan, E. Varathan, V. Subramanian, N. Somanathan and A. B. Mandal, J. Phys. Chem. C, 2014, 118, 28084–28094 CAS.
- J. Huang, R. Tang, T. Zhang, Q. Li, G. Yu, S. Xie, Y. Liu, S. Ye, J. Qin and Z. Li, Chem.–Eur. J., 2014, 20, 5317–5326 CrossRef CAS PubMed.
- J. Huang, X. Yang, X. Li, P. Chen, R. Tang, F. Li, P. Lu, Y. Ma, L. Wang, J. Qin, Q. Li and Z. Li, Chem. Commun., 2012, 48, 9586–9588 RSC.
- X. Shen, F. Liang, G. Zhang and D. Zhang, Analyst, 2012, 137, 2119–2123 RSC.
- X. Gu, G. Yang, G. Zhang, D. Zhang and D. Zhu, ACS Appl. Mater. Interfaces, 2011, 3, 1175–1179 CAS.
- J. Mei, N. L. Leung, R. T. Kwok, J. W. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed.
- L. Wang, K. Wang, B. Zou, K. Ye, H. Zhang and Y. Wang, Adv. Mater., 2015, 27, 2918–2922 CrossRef CAS PubMed.
- Q. Qi, J. Qian, X. Tan, J. Zhang, L. Wang, B. Xu, B. Zou and W. Tian, Adv. Funct. Mater., 2015, 25, 4005–4010 CrossRef CAS.
- H. Zhao, Y. Wang, Y. Wang, G. He, M. Xue, P. Guo, B. Dai, Z. Liu and Y. Qi, RSC Adv., 2015, 5, 19176–19181 RSC.
- L. Ding, S. C. Dong, Z. Q. Jiang, H. Chen and L. S. Liao, Adv. Funct. Mater., 2015, 25, 645–650 CrossRef CAS.
- W. Qin, J. W. Lam, Z. Yang, S. Chen, G. Liang, W. Zhao, H. S. Kwok and B. Z. Tang, Chem. Commun., 2015, 51, 7321–7324 RSC.
- X. Zhan, N. Sun, Z. Wu, J. Tu, L. Yuan, X. Tang, Y. Xie, Q. Peng, Y. Dong and Q. Li, Chem. Mater., 2015, 27, 1847–1854 CrossRef CAS.
- H. H. Fang, J. Yang, J. Feng, T. Yamao, S. Hotta and H. B. Sun, Laser Photonics Rev., 2014, 8, 687–715 CrossRef.
- W. Dong, Y. Pan, M. Fritsch and U. Scherf, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 1753–1761 CrossRef CAS.
- M. Gao, L. Wang, J. Chen, S. Li, G. Lu, L. Wang, Y. Wang, L. Ren, A. Qin and B. Z. Tang, Chem.–Eur. J., 2016, 22, 5107–5112 CrossRef CAS PubMed.
- X. Ouyang, X.-L. Li, L. Ai, D. Mi, Z. Ge and S.-J. Su, ACS Appl. Mater. Interfaces, 2015, 7, 7869–7877 CAS.
- M. T. Sajjad, P. P. Manousiadis, H. Chun, D. A. Vithanage, S. Rajbhandari, A. L. Kanibolotsky, G. Faulkner, D. O'Brien, P. J. Skabara and I. D. Samuel, ACS Photonics, 2015, 2, 194–199 CrossRef CAS.
- W. Z. Yuan, P. Lu, S. Chen, J. W. Lam, Z. Wang, Y. Liu, H. S. Kwok, Y. Ma and B. Z. Tang, Adv. Mater., 2010, 22, 2159–2163 CrossRef CAS PubMed.
- T. Förster and K. Kasper, Zeitschrift für Elektrochemie, Berichte der Bunsengesellschaft für physikalische Chemie, 1955, 59, 976–980 Search PubMed.
- N. Zhao, J. W. Lam, H. H. Sung, H. M. Su, I. D. Williams, K. S. Wong and B. Z. Tang, Chem.–Eur. J., 2014, 20, 133–138 CrossRef CAS PubMed.
- J. Luo, Z. Xie, J. W. Y. Lam, L. Cheng, B. Z. Tang, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu and D. Zhu, Chem. Commun., 2001, 1740–1741 RSC.
- T. Zhang, Y. Jiang, Y. Niu, D. Wang, Q. Peng and Z. Shuai, J. Phys. Chem. A, 2014, 118, 9094–9104 CrossRef CAS PubMed.
- Q. Zeng, Z. Li, Y. Dong, C. Di, A. Qin, Y. Hong, L. Ji, Z. Zhu, C. K. Jim, G. Yu, Q. Li, Y. Liu, J. Qin and B. Z. Tang, Chem. Commun., 2007, 70–72 RSC.
- K. A. Upamali, L. A. Estrada, P. K. De, X. Cai, J. A. Krause and D. C. Neckers, Langmuir, 2011, 27, 1573–1580 CrossRef CAS PubMed.
- G. Zhang, J. Lu and C. L. Fraser, Inorg. Chem., 2010, 49, 10747–10749 CrossRef CAS PubMed.
- J. Li, W. Yang, W. Zhou, C. Li, Z. Cheng, M. Li, L. Xie and Y. Li, RSC Adv., 2016, 6, 35833–35841 RSC.
- Z. Zhao, P. Lu, J. W. Y. Lam, Z. Wang, C. Y. K. Chan, H. H. Y. Sung, I. D. Williams, Y. Ma and B. Z. Tang, Chem. Sci., 2011, 2, 672–675 RSC.
- A. Qin, J. W. Lam, F. Mahtab, C. K. Jim, L. Tang, J. Sun, H. H. Sung, I. D. Williams and B. Z. Tang, Appl. Phys. Lett., 2009, 94, 253308 CrossRef.
- E. P. Parrott, N. Y. Tan, R. Hu, J. A. Zeitler, B. Z. Tang and E. Pickwell-MacPherson, Mater. Horiz., 2014, 1, 251–258 RSC.
- J. Chen, B. Xu, X. Ouyang, B. Z. Tang and Y. Cao, J. Phys. Chem. A, 2004, 108, 7522–7526 CrossRef CAS.
- N. L. Leung, N. Xie, W. Yuan, Y. Liu, Q. Wu, Q. Peng, Q. Miao, J. W. Lam and B. Z. Tang, Chem.–Eur. J., 2014, 20, 15349–15353 CrossRef CAS PubMed.
- S. Li, L. He, F. Xiong, Y. Li and G. Yang, J. Phys. Chem. B, 2004, 108, 10887–10892 CrossRef CAS.
- B.-K. An, D.-S. Lee, J.-S. Lee, Y.-S. Park, H.-S. Song and S. Y. Park, J. Am. Chem. Soc., 2004, 126, 10232–10233 CrossRef CAS PubMed.
- Z. Chi, X. Zhang, B. Xu, X. Zhou, C. Ma, Y. Zhang, S. Liu and J. Xu, Chem. Soc. Rev., 2012, 41, 3878–3896 RSC.
- H. Li, X. Zhang, Z. Chi, B. Xu, W. Zhou, S. Liu, Y. Zhang and J. Xu, Org. Lett., 2011, 13, 556–559 CrossRef CAS PubMed.
- P. Chen, R. Lu, P. Xue, T. Xu, G. Chen and Y. Zhao, Langmuir, 2009, 25, 8395–8399 CrossRef CAS PubMed.
- P. Zhang, H. Wang, H. Liu and M. Li, Langmuir, 2010, 26, 10183–10190 CrossRef CAS PubMed.
- H. Zhang, S. Wang, Y. Li, B. Zhang, C. Du, X. Wan and Y. Chen, Tetrahedron, 2009, 65, 4455–4463 CrossRef CAS.
- M.-S. Yuan, X. Du, F. Xu, D.-E. Wang, W.-J. Wang, T.-B. Li, Q. Tu, Y. Zhang, Z. Du and J. Wang, Dyes Pigm., 2015, 123, 355–362 CrossRef CAS.
- Y. Liu, X. Tao, F. Wang, J. Shi, J. Sun, W. Yu, Y. Ren, D. Zou and M. Jiang, J. Phys. Chem. C, 2007, 111, 6544–6549 CAS.
- Y. Liu, X. Tao, F. Wang, X. Dang, D. Zou, Y. Ren and M. Jiang, J. Phys. Chem. C, 2008, 112, 3975–3981 CAS.
- T. Zhou, F. Li, Y. Fan, W. Song, X. Mu, H. Zhang and Y. Wang, Chem. Commun., 2009, 3199–3201 RSC.
- J. Shi, N. Chang, C. Li, J. Mei, C. Deng, X. Luo, Z. Liu, Z. Bo, Y. Q. Dong and B. Z. Tang, Chem. Commun., 2012, 48, 10675–10677 RSC.
- F. Xu, H. Wang, X. Du, W. Wang, D.-E. Wang, S. Chen, X. Han, N. Li, M.-S. Yuan and J. Wang, Dyes Pigm., 2016, 129, 121–128 CrossRef CAS.
- S. K. Lee, W. J. Yang, J. J. Choi, C. H. Kim, S.-J. Jeon and B. R. Cho, Org. Lett., 2005, 7, 323–326 CrossRef CAS PubMed.
- Y. Kawamura, H. Sasabe and C. Adachi, Jpn. J. Appl. Phys., 2004, 43, 7729–7730 CrossRef CAS.
- G. Sheldrick, SHELX-97, Gottingen University, Germany, 1997 Search PubMed.
- E. Zojer, A. Pogantsch, E. Hennebicq, D. Beljonne, J.-L. Brédas, P. S. de Freitas, U. Scherf and E. J. List, J. Chem. Phys., 2002, 117, 6794–6802 CrossRef CAS.
- The CCDC no. of compound ATPF: 1471518.†.
Footnote |
| † Electronic supplementary information (ESI) available: Single crystal, theoretical calculation and additional spectra. CCDC 1471518. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra13102f |
| This journal is © The Royal Society of Chemistry 2016 |