
Bisanthra-thianthrene: synthesis, structure and oxidation properties†
Abstract
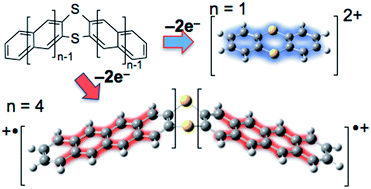
A bisanthra-thianthrene has been synthesized and the oxidation property was studied to show that the bis(tetracene radical cation) linked by a dithiin ring is more stable than the π-expanded dithianonacene dication.
 Please wait while we load your content...
Please wait while we load your content...

