
Cu–Pd/γ-Al2O3 catalyzed the coupling of multi-step reactions: direct synthesis of benzimidazole derivatives
Feng Feng,
Jia Ye,
Zheng Cheng,
Xiaoliang Xu,
Qunfeng Zhang*,
Lei Ma,
Chunshan Lu and
Xiaonian Li*
Institute of Industrial Catalysis of Zhejiang University of Technology, State Key Laboratory Breeding Base of Green Chemistry Synthesis Technology, Hangzhou, 310032, China. E-mail: zhangqf@zjut.edu.cn; xnli@zjut.edu.cn; Fax: +86-571-88320930; Tel: +86-571-88320836 Tel: +86-571-88320002
First published on 22nd July 2016
Abstract
The coupling of multi-step reactions catalyzed by a heterogeneous catalyst is an important path to accomplish some unconventional chemical transformations. Since the starting materials generated from previous steps were adsorbed on the catalyst, the activation energy of following steps was largely decreased, and thus the reaction conditions were more mild and environmental friendly. Catalyzed by a multifunctional Cu–Pd/γ-Al2O3 catalyst, the transfer hydrogenation and successive cyclization coupling reaction from o-nitroaniline and alcohol to afford benzimidazole derivatives in high yield was realized. The catalyst could be reused several times without loss of activity. The synergies of reforming hydrogenation of Cu–Pd bimetal and support acidity of γ-Al2O3 were responsible for this catalytic transformation.
Introduction
The syntheses of complex organic compounds often require multi-step reactions, in which harsh reaction conditions and complex catalysts were frequently used in several steps. In recent decades, use of easily recyclable solid acid replacing liquid acid in the acid-catalyzed reaction was largely developed and considered as a major goal of green chemistry.1 The reactants generated from previous steps were in situ adsorbed on the catalyst surface, which reduced the activation energy for the subsequent steps of the catalyst acidic requirements (Fig. 1). | ||
| Fig. 1 Possible mechanism of coupling reaction. | ||
The gas–liquid–solid three-phase catalytic hydrogenation for the reduction of nitro compounds to amines is an important and conventional technology.2 The solvent, hydrogen, and noble metal catalyst are essential in this catalytic transformation. Although this method is effective, the main drawback is high pressure and temperature, equipment requirements, and at the same time transfer and transport of hydrogen is tedious and dangerous. Recently Dumesic3–8 reported a new hydrogen production method based on transition or noble metal-catalyzed aqueous-reforming technology. Thus, this liquid–solid two phase catalytic transfer hydrogenation provides an efficient route to replace the three-phase catalytic hydrogenation for certain reactions. Use of in situ produced and adsorbed hydrogen on catalyst, nitro compounds are successfully reduced to amines9,10 by this method. And by tuning the acid–base and surface characteristics of support, the multi-step successive coupling reactions of adsorbed intermediates on catalyst may be realized. According to these requirements, modified multifunctional heterogeneous catalyst is an important means to realize this kind of coupling reactions. Based on these, recently the direct reductive N-alkylation of nitrobenzene catalyzed by Raney Ni11 and synthesis of quinoline derivatives from nitrobenzene and alcohol catalyzed by Cu–Pd/γ-Al2O3 was successfully established12 by our group.
Heterocycles are ubiquitous intermediates for the synthesis of pharmaceuticals,13–15 dyes,16,17 organic functional materials.18,19 Among these, the five-membered nitrogen heterocycles, especially benzimidazole derivatives, are widely applied in fungicidal20 and pharmaceutical.13–15 Normally, the condensation of o-phenylenediamine with acid derivatives or o-phenylenediamine with aldehydes in the presence of an appropriate oxidant is a typical way for the synthesis of benzimidazole.21–24 However, the traditional methods mostly suffer from use of strong inorganic acid, formation of large amount of waste, difficult recycling of catalyst, and low atom economy.
Comply with the green chemistry trend, heterogeneous catalytic systems25–37 and microwave technology26,31,38,39 were applied for the synthesis of benzimidazoles recently, such as using P–Mo or P–W heteropoly acid,26,35 modified zeolite,27,28 mesoporous Si–Al material,29 transition metal oxides,30,31 alumina supported potassium fluoride36 and supported noble metal37,39 as catalysts, and using air or oxygen as oxidant.40,41 For instance, Gadekar27 reported that modified zeolite catalyst gave 83–96% yield of benzimidazoles from o-phenylenediamine and aldehydes. Rathod31 reported that benzimidazoles were obtained in 92–94% yield from o-phenylenediamine and aromatic aldehyde using MoO3/CeO2–ZrO2 as catalyst under solvent free condition. Compared with o-phenylenediamines, o-nitroanilines are more readily available substrates. The reductive cyclization of o-nitroaniline with carbonyl derivatives or alcohols is another pathway for the synthesis of benzimidazole.42–50 The direct continuous catalysis in one pot including dehydrogenation of alcohols, successive reduction of o-nitroaniline and cyclization simplified the synthesis process obviously. Based on this concept, Sun50 reported the synthesis of benzimidazole derivatives using methanol both as solvent and carbon source under supercritical condition over copper-doped porous metal oxide catalysts. However, the yield of benzimidazole and N-methylbenzimidazole were 82% and 11% respectively, and the purify procedure was tedious. Selvam44 reported the one-pot photocatalytic synthesis of disubstituted benzimidazoles from N-substituted 2-nitroanilines or 1,2-diamines with good yield (40–96%) over Pt–TiO2 catalyst using solar and UV-A light. The efficiency and yield of target product were found to be higher in UV light than in solar light.
Herein we wish to disclose a convenient way to synthesize benzimidazole derivatives in moderate to excellent yields from o-nitroaniline and alcohol catalyzed by Cu–Pd/γ-Al2O3. The catalysts are rationally designed by the cooperative catalysis concept, where the metal center acts as dehydrogenation and hydrogenation site while the weak acid site acts for the condensation process.
Results and discussion
Firstly, monometallic or bimetal nanoparticles supported on γ-Al2O3 were synthesized. Subsequently, ethanol and o-nitroaniline was chosen as a model compound to evaluate the catalytic activity of catalyst, and the results were listed in Table 1. Monometallic Pd (5% weight amount) loaded on γ-Al2O3 as a catalyst gave very low conversion and yield (entry 1). However, upon alloying palladium with copper, activity of catalysts improved greatly, and different Pd/Cu ratio results in significant difference in activity or yield. 2-Methylbenzimidazole was selectively obtained in 98.2% yield catalyzed by Cu5–Pd5/γ-Al2O3 (entry 4). Since monometallic Cu/γ-Al2O3 was totally inactive for this reaction (entry 6), the improved performance of Cu–Pd/γ-Al2O3 catalyst can be ascribed to the modification of palladium properties by alloying with copper. Changing the bimetal component to Cu–Ni, Cu–Pt, Fe–Pd, Fe–Pt, Zn–Ni, the conversion and yield are disappointing (entries 7–11). Use of 5% weight amount of Zn and Pd loaded on γ-Al2O3, only moderate conversion and yield were obtained (entry 12). From these, it can be concluded that Ni or Pt show lower dehydrogenation and hydrogenation performance, compared with that of Pd.
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Con. (%) | Yieldb (%) | Selectivity (%) |
| a Reaction conditions: 1 g of catalyst and 5% loading for each metal, 6 g of o-nitroaniline, 100 mL of ethanol, 50 mL of H2O, 453 K, 3.5 MPa, 12 h, 900 rpm.b Checked by HPLC.c 1% loading for Pd. | ||||
| 1 | Pd/γ-Al2O3 | 6.5 | 6.5 | 100 |
| 2 | Cu5–Pd1c/γ-Al2O3 | 23.0 | 21.9 | 95.2 |
| 3 | Cu5–Pd3/γ-Al2O | 69.4 | 68.2 | 98.3 |
| 4 | Cu5–Pd5/γ-Al2O3 | 100 | 98.2 | 98.2 |
| 5 | Cu5–Pd10/γ-Al2O3 | 100 | 95.4 | 95.4 |
| 6 | Cu/γ-Al2O3 | — | — | — |
| 7 | Cu–Ni/γ-Al2O3 | 23.5 | 22.5 | 95.7 |
| 8 | Cu–Pt/γ-Al2O3 | 18.7 | 14.6 | 78.1 |
| 9 | Fe–Pd/γ-Al2O3 | 63.0 | 12.2 | 19.4 |
| 10 | Fe–Pt/γ-Al2O3 | 21.3 | 8.7 | 40.8 |
| 11 | Zn–Ni/γ-Al2O3 | 12.0 | 10.2 | 85.0 |
| 12 | Zn–Pd/γ-Al2O3 | 73.5 | 52.0 | 70.7 |

Then different supports were estimated (Table 2). The 5% weight amount of Cu and Pd loaded on α-Al2O3 or γ-Al2O3 as the catalysts were used to check the effect of different alumina carriers on this reaction. The more acidic γ-Al2O3 support gave better result and yield of 2-methylbenzimidazole could be reached 98.2% (entry 2). The selectivity and yield of 2-methylbenzimidazole are very low when MgO were used as a support, maybe because alkaline MgO support is disadvantage for the condensation of aldehyde and o-phenylenediamine. Use of activated carbon as a support gave slightly high yield (entry 4). TiO2 or CeO2 as the supports gave only moderate selectivity, compared with that of γ-Al2O3 (entries 5–6).
| Entry | Catalyst | Conversion (%) | Yieldb (%) | Selectivity (%) |
|---|---|---|---|---|
| a Reaction conditions: 1 g of catalyst and 5% loading for each metal, 6 g of o-nitroaniline, 100 mL of ethanol, 50 mL of H2O, 453 K, 3.5 MPa, 12 h, 900 rpm.b Checked by HPLC. | ||||
| 1 | Cu–Pd/α-Al2O3 | 73.0 | 42.2 | 57.8 |
| 2 | Cu–Pd/γ-Al2O3 | 100 | 98.2 | 98.2 |
| 3 | Cu–Pd/MgO | 59.5 | 8.5 | 11.7 |
| 4 | Cu–Pd/C | 72.9 | 38.7 | 53.1 |
| 5 | Cu–Pd/TiO2 | 62.8 | 40.3 | 64.2 |
| 6 | Cu–Pd/CeO2 | 95.6 | 70.1 | 73.3 |
In addition, the recovery and reuse of the developed Cu–Pd/γ-Al2O3 catalyst could be achieved by a simple phase separation. The catalyst can be reused for at least 6 times without loss of activity when ethanol and o-nitroaniline were used, as shown in Fig. 2. The conversion of each time is 100% and the yield is 97.3%, 96.2%, 97.5%, 95.3%, 96.2% and 95.8%, respectively. This demonstrated that the component, structure and surface characteristic of catalyst are stable after several times of uses.
 | ||
| Fig. 2 Recycling of Cu–Pd/γ-Al2O3 for the synthesis of 2-methylbenzimidazole. Reaction conditions: 0.8 g of catalyst, 8 g of o-nitroaniline, 120 mL of ethanol, 80 mL of H2O, 453 K, 3.5 MPa, 12 h, 900 rpm. | ||
Catalyst characterization
The prepared catalyst was characterized by different techniques to give its structure information. The physical structure parameters of γ-Al2O3, fresh Cu–Pd/γ-Al2O3 and spent Cu–Pd/γ-Al2O3 (reused for 6 times) were measured and results are listed in Table 3. The EDS showed that the Pd and Cu content of fresh Pd–Cu/γ-Al2O3 was around 5%, which was in accordance with the theoretical loading. And the metal contents of spent catalyst were very close to fresh catalyst, indicating no obvious metal components loss during the reaction. The BET analysis disclosed that the specific surface area of prepared catalyst is slightly increased. The total pore volume and average pore size were reduced, compared with that of γ-Al2O3. And the spent catalyst underwent a very slight loss of surface area and pore volume. TEM and SEM (Fig. 3) results showed that the metal particles were loaded on γ-Al2O3 surface with high dispersion, and the average size of metal particles was about 6.9 nm (based on TEM results). Fig. 4 showed X-ray diffraction patterns of Cu–Pd/γ-Al2O3. The XRD spectrum showed that at 2θ = 41.6°, 48.3° and 71.3°, which were attributed to obvious and weak characteristic diffraction peaks of Cu–Pd bimetal, respectively. The peak of elemental Pd was found at 2θ = 39.8°, while the peak of elemental copper could not be found from the XRD spectrum. From these, it is concluded that there has a little elemental palladium existing on the surface of γ-Al2O3 support, whereas the existence of copper should be as an alloy of Cu–Pd. Compared with the fresh catalyst, no obvious change was observed for the spent catalyst XRD spectrum, which indicated that Cu–Pd/γ-Al2O3 kept constant crystalline phase after reused for 6 times. According to these characterization and recycling test results, it could be confirmed that Cu–Pd/γ-Al2O3 remained stable properties and performances in this one-pot synthesis process. In addition, NH3-TPD analysis revealed that the acidity of prepared catalyst was decreased (Fig. 5). | ||
| Fig. 3 TEM (a) and SEM (b) images of Cu–Pd/γ-Al2O3. | ||
 | ||
| Fig. 4 XRD patterns of (a) γ-Al2O3, (b) Cu–Pd/γ-Al2O3 (Fresh), and (c) Cu–Pd/γ-Al2O3 (Spent). | ||
 | ||
| Fig. 5 NH3-TPD profiles of (a) γ-Al2O3 and (b) Cu–Pd/γ-Al2O3. | ||
Possible reaction mechanism
Based on previous reports44,46,50–53 and experimental results, the possible mechanism is shown as below (Scheme 1). Dehydrogenation of ethanol is well known to be effectively catalyzed by Cu50,54–58 which lead to acetaldehyde and active hydrogen that likely be the key intermediates for successive reaction. Subsequently the transfer hydrogenation reduction of o-nitroaniline with the adsorbed active hydrogen by Pd gave o-phenylenediamine,10,12 which then condensed with acetaldehyde on Lewis or Bronsted acid center to give Schiff base.21 The desired product was obtained after molecular nucleophilic cyclization and dehydrogenation on the surface of Pd–Cu/γ-Al2O3. Recently, the solid acid such as modified zeolite and transition-metal oxides27,28,30,31 was reported to be used as the acid catalyst for this condensation process. However, in this work relative weak acidic γ-Al2O3 could also successfully act as the acid center for the condensation of o-phenylendiamine and acetaldehyde. It may be ascribed to that the in situ formed o-phenylendiamine and acetaldehyde which adsorbed on the catalyst surface lower the requirements of activation energy and the catalyst acidity. Finally, the corresponding benzimidazole product was obtained by dehydrogenation of dihydrobenzimidazole intermediate which would be usually aided by the alumina supported Cu catalyst.59 According to the analysis above, it can be concluded that this coupling reaction system was catalyzed by the synergy effects of Cu–Pd/γ-Al2O3 catalyst's multiple roles including dehydrogenation (Cu), transfer hydrogenation (Pd) centers, and Lewis or Bronsted acid sites of γ-Al2O3. | ||
| Scheme 1 Possible mechanism for the direct synthesis of 2-methylbenzimidazole from ethanol and o-nitroaniline. | ||
Synthesis of derivatives
With the optimized conditions in hand, several types of o-nitroaniline and alcohol derivatives were tried (Table 4). The methyl or methoxyl substituted o-nitroanilines can act as good partners. However yields of the corresponding benzimidazole derivatives were very low, when the chloro or fluoro substituted o-nitroaniline was used as the starting material. The reason may be the nucleophilic of amino group was decreased due to electron-withdrawing ability of chloro or fluoro atom. Methanol, ethanol, n-propanol, n-butanol, benzyl alcohol were all appropriate candidates.
|
|||||
|---|---|---|---|---|---|
| Entry | R1 | R2 | Cat. (g) | Conversion (%) | Yieldb (%) |
| a Reaction conditions: 8 g of o-nitroaniline, 120 mL of alcohol, 80 mL of H2O, 453 K, 3.5 MPa, 12 h, 900 rpm.b checked by HPLC. | |||||
| 1 | H | H | 0.8 | 100 | 96.5 |
| 2 | H | Me | 0.8 | 100 | 97.4 |
| 3 | H | Et | 0.8 | 100 | 96.2 |
| 4 | H | n-Pr | 1.2 | 100 | 98.7 |
| 5 | H | Ph | 0.8 | 100 | 100 |
| 6 | Me | H | 0.8 | 100 | 98.7 |
| 7 | Me | Me | 0.8 | 100 | 97.5 |
| 8 | Me | Et | 0.8 | 100 | 97.5 |
| 9 | MeO | H | 0.8 | 100 | 97.6 |
| 10 | MeO | Me | 0.8 | 100 | 98.5 |
| 11 | MeO | Et | 0.8 | 100 | 96.8 |
| 12 | Cl | H | 0.8 | 15.8 | 9.3 |
| 13 | Cl | Me | 0.8 | 10.5 | 5.8 |
| 14 | F | H | 0.8 | 22.9 | 17.5 |
| 15 | F | Me | 0.8 | 23.8 | 13.9 |

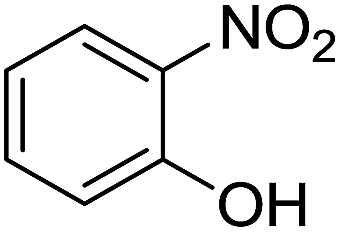
To further establish the general utility of these transformations, o-nitroaniline was replaced with different nitro-compounds for the synthesis of other heterocyclic compounds (Table 5), affording the corresponding benzoxazole, benzothiazole and quinoline compound. But yields of the corresponding heterocyclic compounds were relatively low.







Conclusions
In conclusion, the direct synthesis of benzimidazole from the o-nitroaniline and alcohol was successfully established by tuning the types of metal and support. The combination of Cu and Pd was responsible for the dehydrogenation of alcohol and hydrogenation of o-nitroaniline. The γ-Al2O3 carrier supplied the Lewis or Bronsted acid center to promote the condensation of in situ formed o-phenylenediamine and aldehyde, which gave the desired benzimidazole derivatives after oxidation dehydrogenation. The method described here has the advantage of easily available starting materials, high efficiency, and simple procedure.Experimental
Catalyst preparation
The Cu–Pd/γ-Al2O3 catalyst was prepared by impregnation with precursors of aqueous solution of Cu(NO3)2 (0.05 gmetal/mL) and H2PdCl4 (0.05 gmetal/mL). After γ-Al2O3 support was stirred in distilled water for 10 minutes at 80 °C, H2PdCl4 solution was added dropwise followed by aqueous solution of Cu(NO3)2, and kept the solution at 80 °C for 5 hours. Then pH value was adjusted to 8–9 by adding aqueous NaHCO3. After maintained at this condition for 5 minutes, the solid was filtered and washed to neutral by distilled water, then washed by ethanol, distilled water, successively. The resulting solid was dried in vacuo at 383 K and then calcined at 533 K for 3 hours, finally reduced by H2 at 533 K for 2 hours to give the catalyst.Characterization of catalysts
The bulk composition of the samples was determined via energy-dispersive X-ray spectroscopy (EDS) on Noran VANTAGE-ESI spectrometer. The surface area and porosity analysis of catalysts were measured with N2 physisorption at 77 K on a Micromeritics ASAP 2020. Transmission electron microscope (TEM) analysis was performed with a Tencnai G2 F30 S-Twin instrument operating at 300 kV. Average particle size was calculated by 300 particles which were randomly measured. Scanning electron microscopy (SEM) was analysed on a Hitachi S-4700 instrument at 15 kV. X-ray powder diffraction (XRD) was obtained on a Thermo ARL SCINTAG X' TRA diffractometer using Cu Kα radiation (λ = 0.15406 nm) at 45 kV and 40 mA. The scanning range is from 2θ = 10–80°with a scanning rate of 28 min−1. The diffraction peaks obtained were referred to the JCPDS cards. Prior to each experiment, catalyst (0.075 g) was heated in a flow of He (30 mL min−1) at 533 K for 40 min, followed by cooling to 323 K under He flow. Then, the catalyst was exposed to a flow of 100% NH3 for 30 min. After being purged in He for 60 min, the catalyst was heated linearly at 10 K min−1 to 973 K in a flow of He, and NH3 (m/z = 17) in the outlet gas were analyzed by the mass spectrometer (BELMASS).Synthesis of benzimidazoles
The reaction was performed in a 500 mL stainless steel autoclave, which was charged with 8 g of the nitroaromatic compound,120 mL alcohol, 80 mL deionized water and 0.8 g of the catalyst. The batch reactor was initially purged five times with pure N2 to replace the air in the system. The reactor was then heated to the desired temperature at the desired N2 pressure and at a stirring rate of 900 rpm. After the complete conversion of the reactant, the catalyst was filtered from the mixture for subsequent use, and the corresponding product was separated from the aqueous phase using a separatory funnel. The products were identified by gas chromatography-mass spectrometry (GC-MS, Agilent 5973N) and were analyzed by HPLC using a Baseline C18 column (150 mm × 2.1 nm) with a mobile phase of ethanol–ethyl acetate–water (7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:2 by volume) at a 1.2 mL min−1 constant flow. Quantitative analysis was conducted using the area-normalization method.
:1:2 by volume) at a 1.2 mL min−1 constant flow. Quantitative analysis was conducted using the area-normalization method.
Recycling tests
After a typical catalytic run (8 g o-nitroaniline, 120 mL ethanol, 80 mL H2O, 0.8 g Cu–Pd/γ-Al2O3 catalyst, 453 K, 3.5 MPa, 900 rpm, 12 h), the catalyst was filtered, additionally washed with alcohol and deionized water, and reused after dried in vacuo at 383 K.Acknowledgements
Financial support by the National Natural Science Foundation of China (21406199 and 21476208) is gratefully acknowledged.Notes and references
- H. Hattori, Top. Catal., 2010, 53, 432–438 CrossRef CAS.
- A. M. Tafesh and J. Weiguny, Chem. Rev., 1996, 96, 2035–2052 CrossRef CAS PubMed.
- R. D. Cortright, R. R. Davda and J. A. Dumesic, Nature, 2002, 418, 964–967 CrossRef CAS PubMed.
- G. W. Huber, J. W. Shabaker and J. A. Dumesic, Science, 2003, 300, 2075–2077 CrossRef CAS PubMed.
- R. R. Davada and J. A. Dumesic, Angew. Chem., 2003, 42, 4068–4071 CrossRef PubMed.
- J. W. Shabaker, G. W. Huber and J. A. Dumesic, J. Catal., 2004, 222, 180–191 CrossRef CAS.
- R. R. Davada and J. A. Dumesic, Chem. Commun., 2004, 1, 36–37 RSC.
- R. R. Davda, J. W. Shabaker, G. W. Huber, R. D. Cortright and J. A. Dumesic, Appl. Catal., B, 2005, 56, 171–186 CrossRef CAS.
- L. Zhou, H. Z. Gu and X. H. Yan, Catal. Lett., 2009, 132, 16–21 CrossRef CAS.
- Y. Z. Xiang, Q. Q. Meng, X. N. Li and J. G. Wang, Chem. Commun., 2010, 46, 5918–5920 RSC.
- X. N. Li, J. H. Zhang, Y. Z. Xiang, L. Ma, Q. F. Zhang, C. S. Lu, H. Wang and Y. Bai, Sci. China, Ser. B: Chem., 2008, 38, 27–34 Search PubMed.
- F. Feng, Y. Z. Xiang, X. H. Wang, L. Ma, C. S. Lu, Q. F. Zhang, X. L. Xu and X. N. Li, Sci. China, Ser. B: Chem., 2011, 41, 914–924 Search PubMed.
- W. A. Denny, G. W. Rewcastle and B. C. Baguley, J. Med. Chem., 1990, 33, 814–819 CrossRef CAS PubMed.
- A. R. Porcari, R. V. Devivar, L. S. Kucera, J. C. Drach and L. B. Townsend, J. Med. Chem., 1998, 41, 1252–1262 CrossRef CAS PubMed.
- S. T. Huang, I. J. Hsei and C. Chen, Bioorg. Med. Chem., 2006, 14, 6106–6119 CrossRef CAS PubMed.
- J. A. Asensio and P. Gomez-Romero, Fuel Cells, 2005, 5, 336–343 CrossRef CAS.
- H. Yoshida, R. Nakao, H. Nohta and M. Yamaguchi, Dyes Pigm., 2000, 47, 239–245 CrossRef CAS.
- F. S. Rodembusch, T. Buckup, M. Segala, L. Tavares, R. R. B. Correia and V. Stefani, Chem. Phys., 2004, 305, 115–121 CrossRef CAS.
- J. R. Gong, L. J. Wan, S. B. Lei, C. L. Bai, X. H. Zhang and S. T. Lee, J. Phys. Chem. B, 2005, 109, 1675–1682 CrossRef CAS PubMed.
- S. Sharma, S. Gangal and A. Rauf, Eur. J. Med. Chem., 2009, 44, 1751–1757 CrossRef CAS PubMed.
- M. A. Phillips, J. Chem. Soc., 1928, 1928, 2393–2399 RSC.
- D. W. Hein, R. J. Alheim and J. J. Leavitt, J. Am. Chem. Soc., 1957, 79, 427–429 CrossRef CAS.
- J. J. Vanden Eynde, F. Delfosse, P. Lor and Y. Van Haverbeke, Tetrahedron, 1995, 51, 5813–5818 CrossRef CAS.
- I. Bhatnagar and M. V. George, Tetrahedron, 1968, 24, 1293–1298 CrossRef CAS.
- H. Sharghi, O. Asemani and S. M. H. Tabaei, J. Heterocycl. Chem., 2008, 45, 1293–1298 CrossRef CAS.
- R. Fazaeli and H. Aliyan, Appl. Catal., A, 2009, 353, 74–79 CrossRef CAS.
- L. S. Gadekar, B. R. Arbad and M. K. Lande, Chin. Chem. Lett., 2010, 21, 1053–1056 CrossRef CAS.
- K. W. Ahmad, S. A. Majid and T. Radha, Res. J. Chem. Environ., 2013, 17, 40–45 CAS.
- M. A. Chari, D. Shobha, E. Kenawy, S. S. Al-Deyab, B. V. S. Reddy and A. Vinu, Tetrahedron Lett., 2010, 51, 5195–5199 CrossRef CAS.
- R. V. Shingalapur and K. M. Hosamani, Catal. Lett., 2010, 137, 63–68 CrossRef CAS.
- S. B. Rathod, M. K. Lande and B. R. Arbad, Bull. Korean Chem. Soc., 2010, 31, 2835–2840 CrossRef CAS.
- F. K. Behbahani, P. Ziaei, Z. Fakhroueian and N. Doragi, Monatsh. Chem., 2011, 142, 901–906 CrossRef CAS.
- M. Rekha, A. Hamza, B. R. Venugopal and N. Nagaraju, Chin. J. Catal., 2012, 33, 439–446 CrossRef CAS.
- A. Teimouri, A. N. Chermahini, H. Salavati and L. Ghorbanian, J. Mol. Catal. A: Chem., 2013, 373, 38–45 CrossRef CAS.
- M. M. Heravi, S. Sadjadi, H. A. Oskooie, R. H. Shoar and F. F. Bamoharram, Catal. Commun., 2008, 9, 504–507 CrossRef CAS.
- S. B. Khalili and A. R. Sardarian, Monatsh. Chem., 2012, 143, 841–846 CrossRef CAS.
- V. R. Ruiz, A. Corma and M. J. Sabater, Tetrahedron, 2010, 66, 730–735 CrossRef CAS.
- D. C. Mungra, M. P. Patel and R. G. Patel, Med. Chem. Res., 2011, 20, 782–789 CrossRef CAS.
- L. D. Luca and A. Porcheddu, Eur. J. Org. Chem., 2011, 2011, 5791–5795 CrossRef.
- S. Park, J. Jung and E. J. Cho, Eur. J. Org. Chem., 2014, 2014, 4148–4154 CrossRef CAS.
- R. Jain, D. D. Agarwal, P. K. Sahu, D. T. Selvam, Y. Sharma, R. Gupta and A. Prakash, Med. Chem. Res., 2013, 22, 1788–1794 CrossRef CAS.
- H. Wang, R. E. Partch and Y. Li, J. Org. Chem., 1997, 62, 5222–5225 CrossRef CAS.
- M. P. Surpur, P. R. Singh, S. B. Patil and S. D. Samant, Synth. Commun., 2007, 37, 1375–1379 CrossRef CAS.
- K. Selvam and M. Swaminathan, Tetrahedron Lett., 2011, 52, 3386–3392 CrossRef CAS.
- M. Annadhasan, K. Selvam and M. Swaminathan, Synth. Commun., 2012, 42, 1500–1508 CrossRef CAS.
- N. A. Weires, J. Boster and J. Magolan, Eur. J. Org. Chem., 2012, 2012, 6508–6512 CAS.
- G. Li, J. Wang, B. Yuan, D. F. Zhang, Z. Y. Lin, P. Li and H. H. Huang, Tetrahedron Lett., 2013, 54, 6934–6936 CrossRef CAS.
- L. Tang, X. F. Guo, Y. Yang, Z. G. Zha and Z. Y. Wang, Chem. Commun., 2014, 50, 6145–6148 RSC.
- J. Chen, S. J. Huang, J. Lin and W. P. Su, Res. J. Chem. Environ., 2014, 470, 1–7 CrossRef CAS PubMed.
- Z. H. Sun, G. Bottari and K. Barta, Green Chem., 2015, 17, 5172–5181 RSC.
- Y. Shiraishi, Y. Sugano, S. Tanaka and T. Hirai, Angew. Chem., Int. Ed., 2010, 49, 1656–1660 CrossRef CAS PubMed.
- A. Saha and B. Ranu, J. Org. Chem., 2008, 73, 6867–6870 CrossRef CAS PubMed.
- P. Ghosh and A. Mandal, Catal. Commun., 2011, 12, 744–747 CrossRef CAS.
- D. J. Elliot and F. Pennella, J. Catal., 1989, 119, 359–367 CrossRef.
- K. Inui, T. Kurabayashi and S. Sato, J. Catal., 2002, 212, 207–215 CrossRef CAS.
- Y. J. Tu and Y. W. Chen, Ind. Eng. Chem. Res., 1998, 37, 2618–2622 CrossRef CAS.
- Y. J. Tu and Y. W. Chen, Ind. Eng. Chem. Res., 2001, 40, 5889–5893 CrossRef CAS.
- F. Wang, R. J. Shi, Z. Q. Liu, P. J. Shang, X. Y. Pang, S. Shen, Z. C. Feng, C. Li and W. J. Shen, ACS Catal., 2013, 3, 890–894 CrossRef CAS.
- D. Damodara, R. Arundhathi and P. R. Likhar, Adv. Synth. Catal., 2014, 356, 189–198 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2016 |