
Confined crystallization of poly(butylene succinate) intercalated into organoclays: role of surfactant polarity
Jian Tengab,
Ben Niua,
Liang-Qing Zhanga,
Xu Jib,
Ling Xu*a,
Zheng Yana,
Jian-Hua Tangb,
Gan-Ji Zhong*a and
Zhong-Ming Lia
aCollege of Polymer Science and Engineering, State Key Laboratory of Polymer Materials Engineering, Sichuan University, Chengdu 610065, People's Republic of China. E-mail: ganji.zhong@scu.edu.cn; lingxu@scu.edu.cn
bCollege of Chemical Engineering, Sichuan University, Chengdu 610065, People's Republic of China
First published on 12th July 2016
Abstract
Understanding crystalline morphology and crystallization kinetics of PBS in the presence of a nanoclay is crucial to reveal the relationship between properties, morphology, as well as the processing of PBS/clay nanocomposites. In this work, two types of organoclay with slightly different polarity are homogeneously dispersed in PBS by melt intercalation. Interestingly, the crystallization kinetics of poly(butylene succinate) (PBS) are seriously affected by this slightly different polarity of the organic modifier grafted nanoclay. During isothermal crystallization of PBS in the presence of an organoclay, the crystallization behavior is significantly confined by an organoclay with slightly stronger polarity compared to that of an organoclay with a relatively weak polarity. Further, the nucleation density and crystallinity of PBS in the presence of 20 wt% of an organoclay with slightly stronger polarity are separately decreased 48.8% and 13.4% compared to neat PBS, while another organoclay with weak polarity facilitates nucleation and has a negligible influence on crystallinity. This study offers a new insight into the effect of the organic modifier polarity of a nanoclay on the confined crystallization of PBS, which provides significant guidance for fabricating a high barrier PBS film by using confined crystals as reported in our previous work.
1. Introduction
In the past decade, biodegradable polymers, which can be completely degraded in soil and do not release any noxious components, have received extensive attention because of finite petroleum resources and growing environmental issues. Among various biodegradable polymers, poly(butylene succinate) (PBS) is a commercially available and widely investigated polyester,1,2 which possesses outstanding ductility, good melt processability, promising heat resistance, and tensile strength comparable to polypropylene. Furthermore, PBS/clay nanocomposites often present enhanced properties compared to micro-scale fillers, such as higher modulus,3,4 enhanced tensile strength,5 and better barrier properties,6 which could shed light on the realization of high performance of PBS and finally the enlargement of its applications. To achieve commercialization, understanding crystalline morphology and crystallization kinetics of PBS in the presence of nanoclay is crucial to reveal the relationship between properties, morphology, as well as processing, which already draw some attentions.7–9 For instance, Im et al. studied the influence of clay surface modified by urethane group on the crystallization of PBS,8 and Ray et al. well reviewed the recent progress on crystallization of PBS/nanoclay nanocomposites.9Nanoclay has been acknowledged as highly effective reinforced filler for polymeric matrix, due to its extraordinarily high aspect ratio and inherently superior mechanical properties.10 Thus, incorporating nanoclay into PBS matrix seems to be a versatile strategy for fabricating high performance PBS/nanoclay nanocomposites. Moreover, complete exfoliation and uniform dispersion of nanoclay in the PBS matrix is the prerequisite for improved properties.11–14 Nevertheless, raw clays are hydrophilic, leading to lower degree of exfoliation and inferior molecular interaction with polymer matrix, and thus poor performance. Therefore, various surfactants must be intercalated into the interlayers of clays to improve the dispersion of nanoclays in polymer matrix. The type and nature of surfactant agents are key to affect the dispersion and exfoliation of nanoclay in polymeric matrix.15–17 For instance, Shibata et al. found that protonated ammonium cations composed of longer fatly chain groups are more effective for the intercalation of nanoclays, which leading to better dispersion and thus enhanced mechanical properties of PBS/clay nanocomposites.3
With regard to crystallization, different kinds of organoclay are critical to polymer crystallization behavior, the organic agent in the surface of nanoclay may interact with polymer chains due to polar groups (i.e. the polarity of surfactant), resulting in nucleation density or crystal growth with great difference during crystallization. However, it is still unclear how the surfactant polarity in nanoclay affects the crystallization of PBS. Furthermore, in our previous work, we studied the crystallization behavior of PBS in the presence of clays at wide range of loadings. A novel crystallization behavior was observed, i.e. high loadings of clays had rigorous spatial confinement effect on PBS crystallization.18 And it further revealed that spatial confinement effect played a crucial role in improving the gas barrier performance of PBS film by interdicting permeation paths and confining the diffusion of gas molecules.19 The objective of this work is to investigate the effect of surfactant polarity on the confined crystallization of PBS, two types of surfactants with similar architecture but with different polar groups are chosen to fabricate PBS/clay nanocomposites. It is found that crystallization kinetics of PBS varies with the difference of surfactant polarity due to the interaction between PBS and organoclay. Furthermore, the nucleation density, spherulite growth rate and crystallinity of PBS/organoclay were intensively identified by polarizing optical microscopy (POM), differential scanning calorimetry (DSC), X-ray diffraction (XRD), scanning electron microscope (SEM).
2. Experimental section
2.1. Materials
PBS (model bionolle #1001MD; melt flow rate = 1.6 g/10 min (190 °C, 2.16 kg); weight-average molecular = 2.2 × 105 g mol−1, number-average molecular = 1.1 × 105 g mol−1) was purchased from Showa Denko (Japan). Two types of organically grafted clays (I.28E and I.34CN) were supplied by Nanocor Corporation (America). The chemical structure of modifier and its content can be seen in Table 1.| Clay type | Chemical structure of modifier | Content of modifier (wt%) |
|---|---|---|
| I.28E | 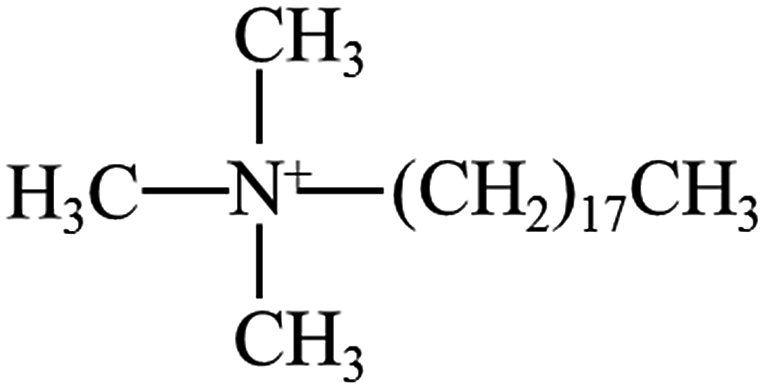 |
28–30 |
| I.34TCN |  |
30–32 |
2.2. Preparation of PBS/organoclay nanocomposites
PBS and clay were dried before use under vacuum for 24 h at 80 °C and 105 °C for 10 h, respectively. Melt blending of PBS and organoclay was carried out in a torque rheometer (XSS-300) with 50 rpm at 150 °C for 10 min. A series of organoclay/PBS were prepared, with various organoclay loadings of 1, 5 and 20 wt%. For brevity, the nanocomposites of PBS/1.28E were encoded as PBS/I.28E1, PBS/I.28E5 and PBS/I.28E20, while PBS/I.34TCN nanocomposites were encoded as PBS/I.34TCN1, PBS/I.34TCN5 and PBS/I.34TCN20. After compounding, all specimens were fabricated into 10 μm thick films and sheets with a diameter of 25 mm and a thickness of 1 mm by the hot pressing, and the temperature, compaction pressure and holding time were 150 °C, 10 MPa and 5 min, respectively.3. Characterizations
3.1. Measurement of crystallization behavior of PBS/organoclay nanocomposites
To investigate crystallization behavior of PBS/organoclay, the nonisothermal and isothermal crystallization process of PBS/organoclay nanocomposites were characterized using a differential scanning calorimetry (DSC, Q2000, TA instrument, USA). Under N2 atmosphere, samples of neat PBS and the nanocomposites (around 5–6 mg) in aluminum pot were heated from ambient temperature to 150 °C at a heating rate of 20 °C min−1 and maintained for 5 min to eliminate thermal history, then cooled to 40 °C for nonisothermal crystallization at a cooling rate of 10 °C min−1 or cooled to 100 °C for isothermal crystallization at a cooling rate of 30 °C min−1. Isothermal crystallization kinetics of PBS/organoclay nanocomposites was stated by classical Avrami equation:20,21
ln[−ln(1 − Xt)] = n![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) lnt + lnk lnt + lnk
| (1) |
Crystallization half-time (t1/2), which could reflect the crystallization rate, was given by:
|
t1/2 = [ln2/k]1/n
| (2) |
Crystalline morphology observation was performed using a polarized optical microscope (POM, BX51, Olympus Co., Tokyo, Japan) equipped with a hot stage (CSS-450, Linkam Scientific Instruments Ltd, UK). The samples were first preserved in molten state at 150 °C for 5 min and then quenched to 100 °C for isothermal crystallization at a rate of 30 °C min−1. In another word, all heat treatment procedure of nanocomposites in hot state was same as DSC measurements. The number of nucleation site was measured by employing an Image-Pro Plus 6.0 software, and the nucleation density was calculated by site numbers per cm−2.
3.2. Observation of clay in matrix and PBS crystallization
The morphology of clay in PBS was investigated on cross-section of films by field emission scanning electron microscopy (FE-SEM, S4800, Hitachi, Japan). The hot-pressed PBS/organoclay nanocomposites were frozen and cryo-fractured directly in liquid nitrogen. Subsequently, the smooth surface was sprayed a layer of thin gold.Rheological measurements of PBS and PBS/organoclay nanocomposites were performed using a TAAR2000EX rotational rheometer (TA Instruments, New Castle, DE) with a parallel plate geometry (diameter = 20 mm), and the gap of parallel plate was 0.7 mm. Before data acquisition, the sample was maintained for 5 min at 150 °C and then frequency sweeps were carried out from 0.01 to 100 Hz at 150 °C with a strain of 1% (within the linear viscoelastic range) for neat PBS and PBS/organoclay nanocomposites.
X-ray diffraction (XRD, Rigaku diffractometer, Japan) was used to characterize the dispersion level of clay in matrix. Nanocomposite sheets were scanned using Cu-Kα radiation (wavelength λ = 1.54 Å) at a rate of 3.6 degree per min. 2D wide-angle X-ray diffraction (2D-WAXD) images were acquired on the beamline BL16B1 (wavelength λ = 1.24 Å), Shanghai Synchrotron Radiation Facility (SSRF, Shanghai, China), to obtain the information of crystal structure and molecular orientation. Previously, the samples were hold at 150 °C for 5 min to erase any thermal history, then quenched to 100 °C for isothermal crystallization. Namely, the specimens were subjected to identical thermal history as described in DSC measurement. The crystallinity (Xc) of PBS can be estimated by:
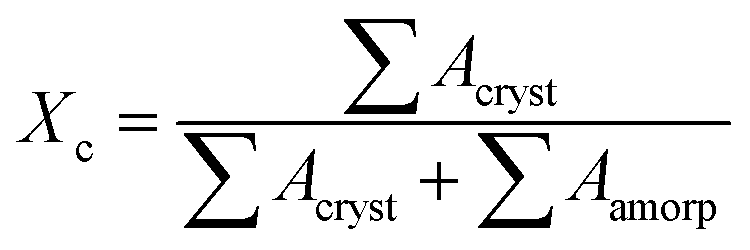 | (3) |
4. Results and discussion
4.1. Nonisothermal and isothermal crystallization behavior of PBS with different organoclays
Nonisothermal crystallization behavior of neat PBS, PBS/I.34TCN and PBS/I.28E are investigated, Fig. 1 shows the DSC cooling curves of PBS, PBS/I.34TCN and PBS/I.28E nanocomposites from equilibrated melt state at 150 °C with a cooling rate of 10 °C min−1, and the crystallization parameters are summarized in Table 2. As we can see, both nonisothermal crystallization temperatures (Tc) of PBS/I.28E and PBS/I.34TCN shift to lower temperature with the increase of organoclay loading, indicating that the PBS crystallization is confined by organoclay and similar to other polymer/montmorillonite nanocomposites.22,23 From Table 2, the initial crystallization temperature (T0), the crystallization temperature (Tc) and the crystallization enthalpy (ΔHc) of PBS are decreased with increasing organoclay loading, but the specific values of PBS/I.34TCN are lower than that of PBS/I.28E at the same mass fraction of organoclay. For example, the Tc of PBS/I.34TCN20 is 80.1 °C, revealing a 0.7 °C decrease compared to PBS/I.28E20 (80.8 °C), manifesting that the nonisothermal crystallization behavior of PBS/I.34TCN is relatively confined more obviously compared to PBS/I.28E. And are the isothermal crystallization of PBS/I.34TCN and PBS/I.28E are similar to?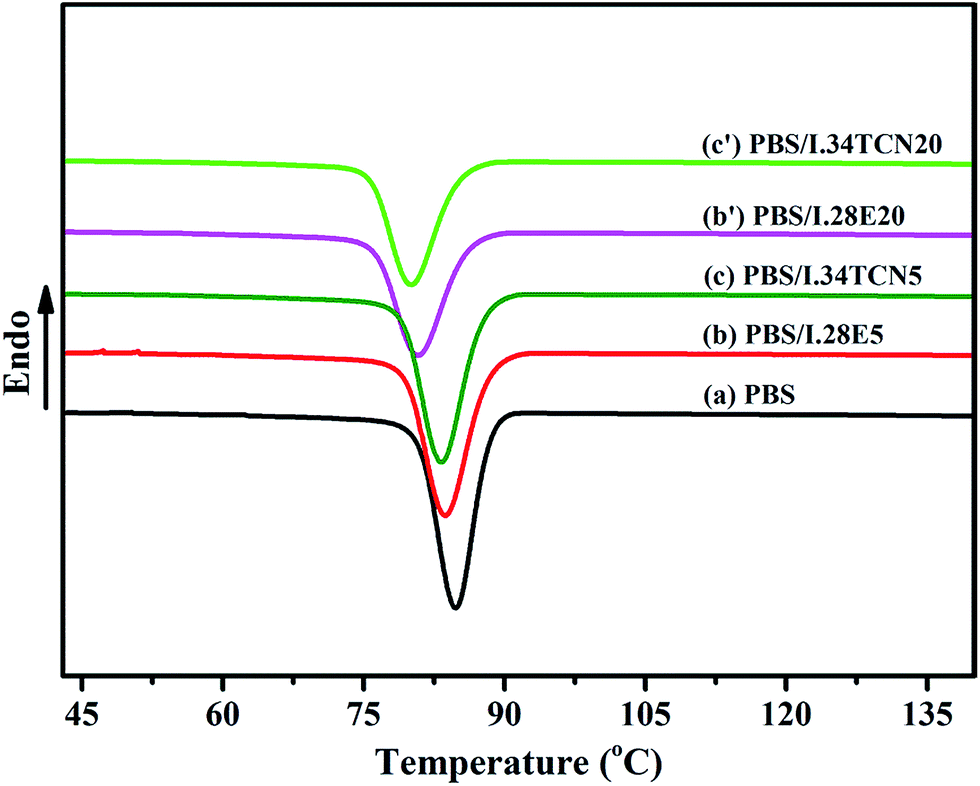 | ||
| Fig. 1 DSC cooling curves of (a) neat PBS; (b)–(b′) PBS/I.28E with increasing organoclay loading; (c)–(c′) PBS/I.34TCN with increasing loading. | ||
| Parameters | PBS | PBS/I.28E5 | PBS/I.34TCN5 | PBS/I.28E20 | PBS/I.34TCN20 |
|---|---|---|---|---|---|
| T0 (°C) | 88.4 | 88.3 | 87.7 | 85.9 | 85.1 |
| Tc (°C) | 84.8 | 83.7 | 83.2 | 80.8 | 80.1 |
| ΔHc (J g−1) | 67.6 | 64.2 | 63.8 | 51.0 | 49.8 |
The isothermal crystallization behavior of PBS/I.28E and PBS/I.34TCN nanocomposites are dramatically different, reflecting by that of half-crystallization time (obtained by DSC). Fig. 2 shows the thermograms of neat PBS and PBS/organoclay nanocomposites during isothermal crystallization at 100 °C. Obviously, isothermal crystallization behaviors of PBS/I.34TCN and PBS/I.28E are very different from nonisothermal crystallization, PBS crystallization is dramatically confined by I.34TCN, while I.28E is not. It can be seen that tmax (the crystallization time corresponding exothermic peak, representing the crystallization rate)20 of PBS/I.28E nanocomposites are reduced compared to that of neat PBS. This phenomenon has been widely reported that nanoclay can act as effective nucleating agent.24,25 With the increased content of I.28E clay in PBS, a spatial confined effect on crystallization occurs. A similar behavior of crystallization has also been reported, a small amount of organoclay in nylon 6 nanocomposites results in a shorter crystallization time compared to neat sample, while higher organoclay loading lengthens the confined crystallization.26 Similarly, tmax of PBS/I.34TCN nanocomposite is significantly shifted toward high value with increasing organoclay content, indicating crystallization rate is remarkably decreased by I.34TCN. But the extent of confined effect is different between two types of nanoclays. For example, tmax of PBS/I.34TCN5 nanocomposite is higher than that of neat PBS and PBS/I.28E5, respectively.
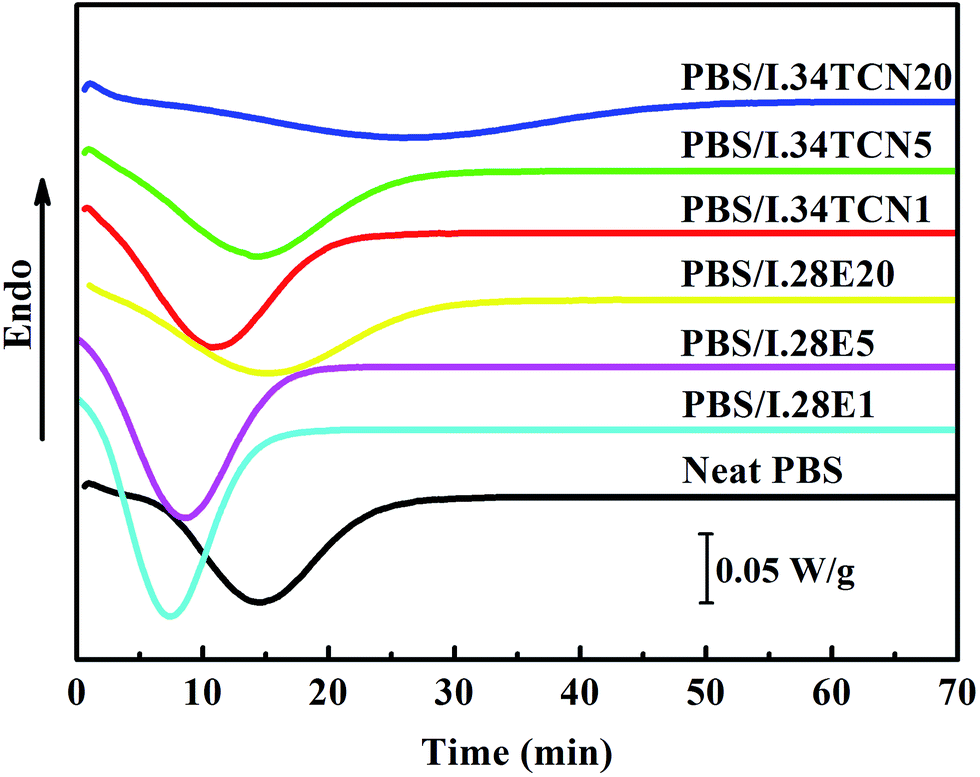 | ||
| Fig. 2 Isothermal crystallization behavior of neat PBS, PBS/I.34TCN and PBS/I.28E nanocomposites at 100 °C. | ||
To distinguish the difference of isothermal crystallization kinetics between PBS/I.34TCN and PBS/I.28E nanocomposites, a series of half crystallization time (t1/2) of PBS/I.28E and PBS/I.34TCN nanocomposites have been calculated by eqn (1) and (2) and plotted in Fig. 3. The varied values of t1/2 with organoclay content manifest a very interesting crystallization behavior of PBS/organoclay nanocomposites. Two transition stages of crystallization kinetics are obviously observed. As the clay content is less than 1 wt%, t1/2 of both nanocomposites decreases and attains its minimum at a loading of 1 wt%. And a significant increase of t1/2 could be obtained after the clay content of PBS is higher than 1 wt%. In other words, crystallization rate of PBS reaches a maximum at around 1 wt% of organoclay loading. For PBS/I.28E, the t1/2 value is decreased at first and then increases with the increase of organoclay loading. And another turning-point appears at around 18 wt% of I.28E organoclay loading (predict from the t1/2 curve as a function of clay), the crystallization rate of PBS/I.28E with lower than 18 wt% of organoclay is enhanced compared to neat PBS and confined after increasing organoclay loading higher than 18 wt%. Similar crystallization behavior has already been widely reported.26,27 At low filler content in nanocomposites, interfaces of matrix-filler act as heterogeneous nucleating sites, greatly increasing crystallization rate. With increasing content, dense fillers confine the diffusion of polymer chains to nucleating sites, hence, reducing crystallization rate. But for PBS/I.34TCN, a similar turning-point appears at 5 wt%. Here we observe that the crystallization of PBS has been confined by I.34TCN clay more obviously than that of I.28E. An obvious tendency can be observed, t1/2 of PBS/I.34TCN nanocomposites is always higher than PBS/I.28E at the same clay loading e.g., the half crystallization time of PBS/I.34TCN1 is 10.9 min, while the value of PBS/I.28E is 7.5 min. The crystallization rate of PBS/I.34TCN nanocomposites is always slower than PBS/I.28E, but how does organoclay affect the ability to PBS crystallization?
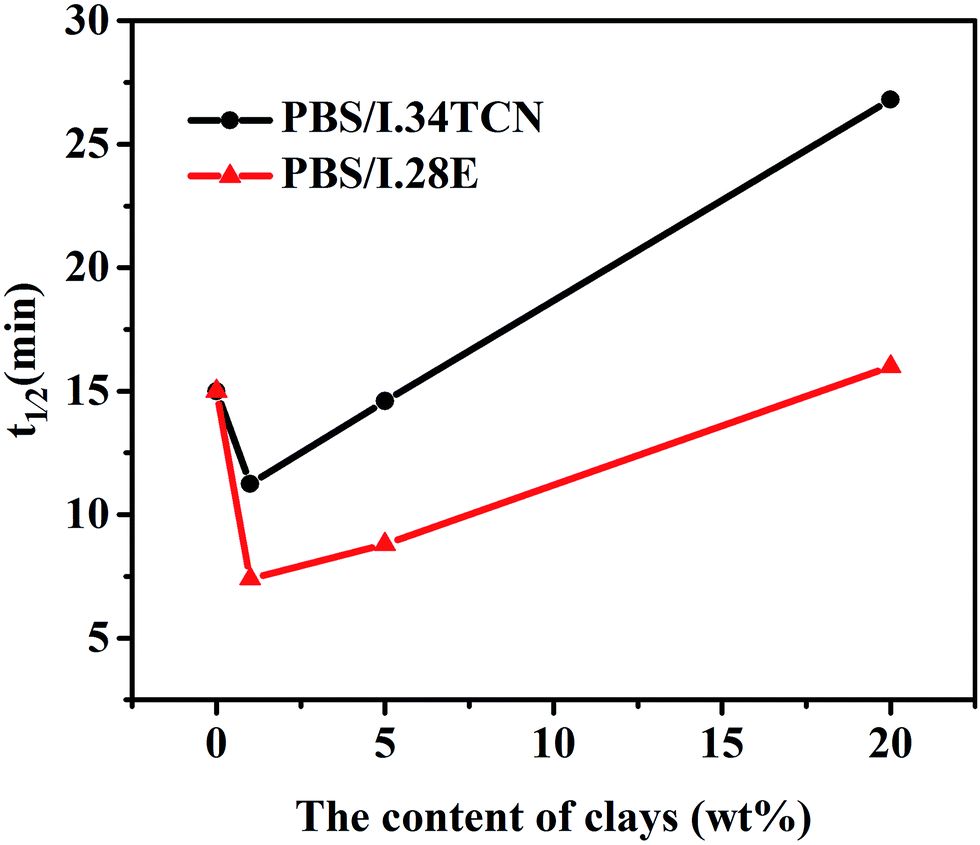 | ||
| Fig. 3 Half-crystallization time of PBS/I.34TCN and PBS/I.28E nanocomposites during isothermal crystallization at 100 °C. | ||
As is well known, polymer crystallization consists of two stages: nucleation and growth, deciding the whole crystallization rate of polymer. It is generally recognized that the nanoclay act as nucleating agent, which could induce nuclei and accelerate crystallization rate.28–31 During the early period of polymer crystallization, large aspect ratio and high specific area of nanoclay act as an effective nucleating template for chains to landscape, resulting in enhancing crystallization rate. The significant crystallization rate difference of PBS/I.34TCN and PBS/I.28E leads us to further explore the formation mechanism on PBS crystalline morphology induced by organoclay.
POM has been employed to observe the overall crystal growth process, and the results are shown in Fig. 4. It is specifically noted that the nucleation site of PBS/I.34TCN nanocomposites appeared in POM (Fig. 4(b–d)) gradually decreases with increasing organoclay content after isothermal crystallization at 100 °C for 8 min. Apparently, the nucleation density of PBS/organoclay nanocomposites is lower than the neat one (Fig. 4(a)), significantly minished by I.34TCN clay addition. As shown in Fig. 5, the nucleation density of PBS/I.34TCN1 is 1.77 × 105 cm−2, decreasing by 48.84% compared to neat PBS (3.46 × 105 cm−2), and dropping further with the increasing content of I.34TCN. While nucleation density of PBS/I.28E is obviously higher than that of neat PBS, e.g., PBS/I.28E20 (5.08 × 105 cm−2) is 46.82% higher than that of neat PBS. It clearly reflects that the I.34TCN may act as a suppressive template to increase energy barrier and to prevent PBS chain fold form nuclei. In clear contrast, I.28E clay obviously increases the nucleation density of PBS spherulite compared with the neat one. It indicates that this type of organoclay usually provides more sites for nucleation or this filler is considered as heterogeneous nucleating agents.32–34 The clay modified by different types of surfactant probably generates a distinct interaction between PBS and organoclay. Due to stronger interaction between PBS and I.34TCN, nucleation activity of PBS on matrix-filler interface is not high. Hence, the nucleation sites are rarely detected or almost none. Meanwhile, the PBS chains motion is restricted by the spatial confinement of I.34TCN and higher concentration of clay, resulting in further attenuated nucleation efficiency. However, weak interaction in PBS/I.28E makes nucleation effortless to generate and expedite PBS spherulitic growth. Therefore, it is no doubt that I.28E serves as efficient nucleating agent to increase nucleation density. The function of surfactant of organoclay among PBS/organoclay nanocomposites can be further demonstrated by the following explorations.
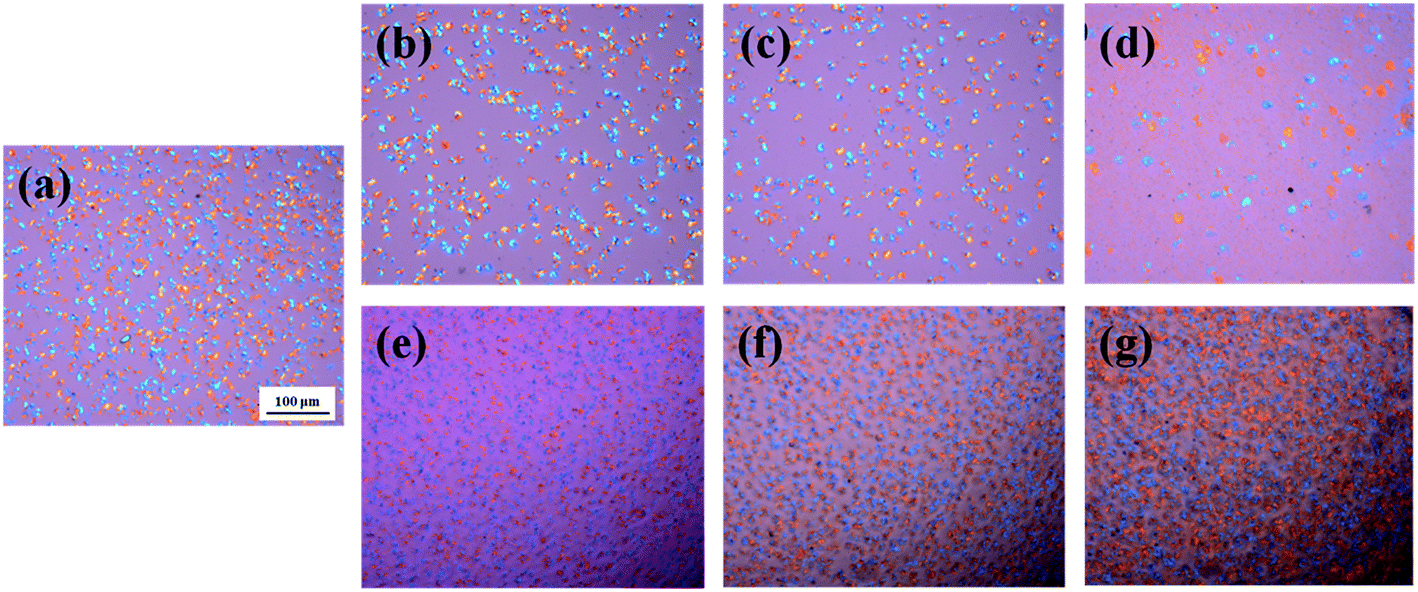 | ||
| Fig. 4 POM images of PBS/organoclay containing I.28E and I.34TCN isothermally crystallized at 100 °C for 8 min: (a) neat PBS, (b) PBS/I.34TCN1, (c) PBS/I.34TCN5, (d) PBS/I.34TCN20, (e) PBS/I.28E1, (f) PBS/I.28E5, (g) PBS/I.28E20. | ||
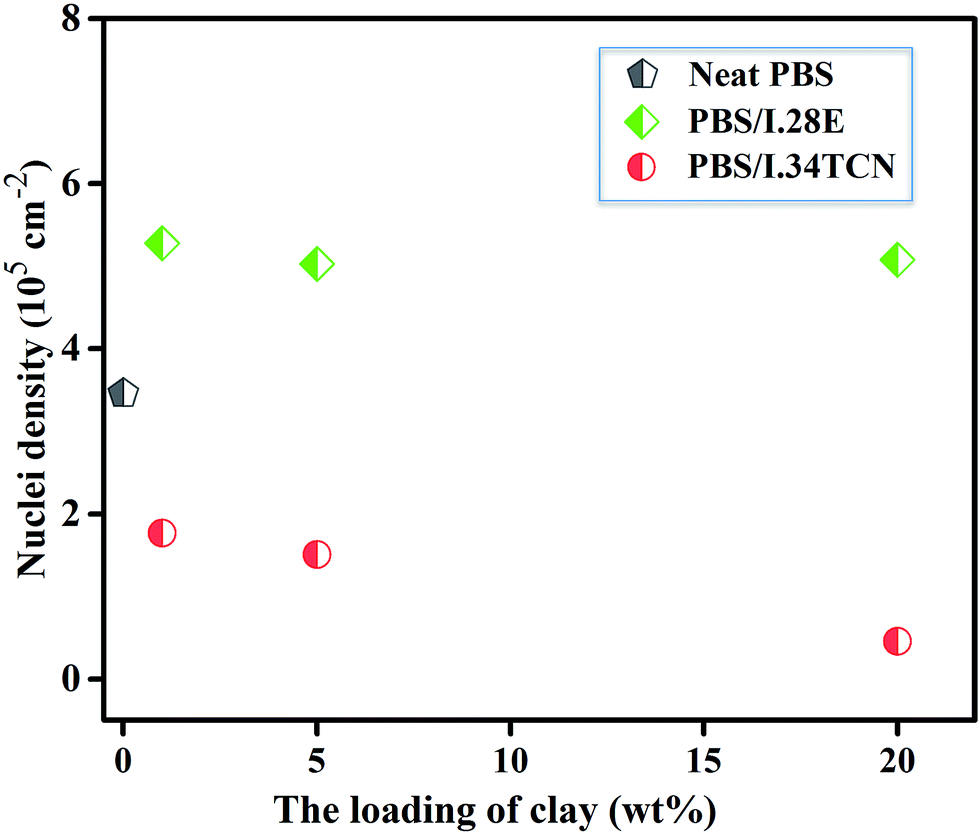 | ||
| Fig. 5 Nucleation densities of neat PBS, PBS/I.28E and PBS/I.34TCN nanocomposites isothermally crystallized at 100 °C for 8 min. | ||
4.2. The dispersion of I.34TCN and I.28E clay in PBS
In this work, the interaction in PBS/organoclay affects the nucleation density and spherulitic growth of PBS, and the dispersion of organoclay is also influenced by the interaction, probably resulting in different crystallization behavior. Therefore, attempts have been made to elucidate the effect of polarity interaction on organoclay dispersion in PBS using XRD and SEM. The XRD curves of various pure organoclay and its nanocomposite are shown in Fig. 6. As we observed that the diffraction peak of (001) plane of pure I.34TCN appears at 4.65°, showing that organoclay layer-to-layer distance is 19.2 Å (according to Bragg equation). The layer spacing is further broadened to 28.9 Å when 20 wt% I.34TCN is incorporated into the neat PBS, which illustrates a well intercalation of organoclay in PBS. The same layer spacing variation of organoclay in PBS/I.28E20 is observed, significantly increasing from 25.5 Å (pure I.28E) to 35.6 Å (PBS/I.28E20). Here, it is noted that the layer spacing of PBS/I.28E is larger than that of PBS/I.34TCN, which may supply commodious space for PBS chains easily entrance into organoclay galleries to induce nucleation. But both layer spacing of PBS/I.34TCN and PBS/I.28E are increased around 10 Å compared to pristine organoclay, indicating the extent of intercalation is roughly equivalent. Therefore, in our case, the effect of dispersion on crystallization can be negligible.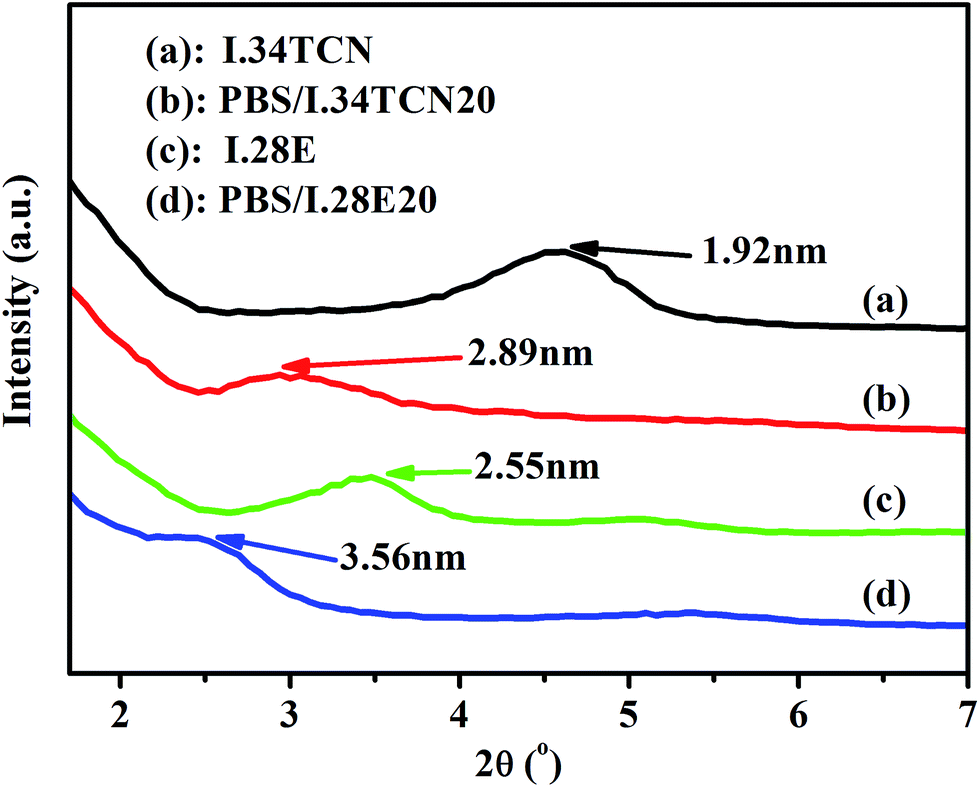 | ||
| Fig. 6 XRD curves of pure I.34TCN, I.28E, PBS/I.34TCN20 and PBS/I.28E20. | ||
The resultant PBS/organoclay nanocomposites were observed by SEM to further estimate the dispersion of organoclay. Fig. 7 presents the SEM micrographs of cross-sections of nanocomposites. Both PBS/I.34TCN and PBS/I.28E display a close-knit texture and no trace of any aggregates of organoclay is discovered, suggesting that the organoclay is efficiently dispersed in PBS. And the fracture surfaces of nanocomposites are very variable, PBS/I.34TCN (Fig. 7(a)) displays a homogeneous and dense texture, while uneven surface emerges in PBS/I.28E (Fig. 7(b)). Perhaps, such different texture architectures are induced by different surfactant. The decreased nucleation density and crystallization rate may due to stronger polar I.34TCN form a forceful interfacial adhesion compared to I.28E under same conditions.
 | ||
| Fig. 7 SEM images of cross-sections of (a) PBS/I.34TCN5 and (b) PBS/I.28E5 nanocomposites. | ||
4.3. Crystal structure of PBS in the nanocomposites with different contents of I.34TCN and I.28E clay
Beyond that, in order to comprehensively ascertain the impact of I.34TCN and I.28E clay on spherulite, the crystal structure of PBS crystallites were further investigated by 2D-WAXD. As shown in insets (2D-WAXD patterns of PBS/I.34TCN5 and PBS/I.28E5) in Fig. 8, similar homogeneous diffraction rings of PBS/organoclay nanocomposites regardless of the content of organoclay are found in diffraction patterns, indicating the formation of isotropic PBS spherulites. There are clear 1D-WAXD curves as illustrated in Fig. 8 after integrating 2D-WAXD patterns. Apparently, two strong peaks appear at 18.1° and 15.7° in all PBS/organoclay and neat PBS, representing lattice planes (110)/(020) of α-form PBS. And there are no other characteristic diffraction peak after isothermal crystallization at 100 °C, suggesting that organoclays, whether I.34TCN or I.28E, and confinement crystallization have no effect on the crystal structure of PBS.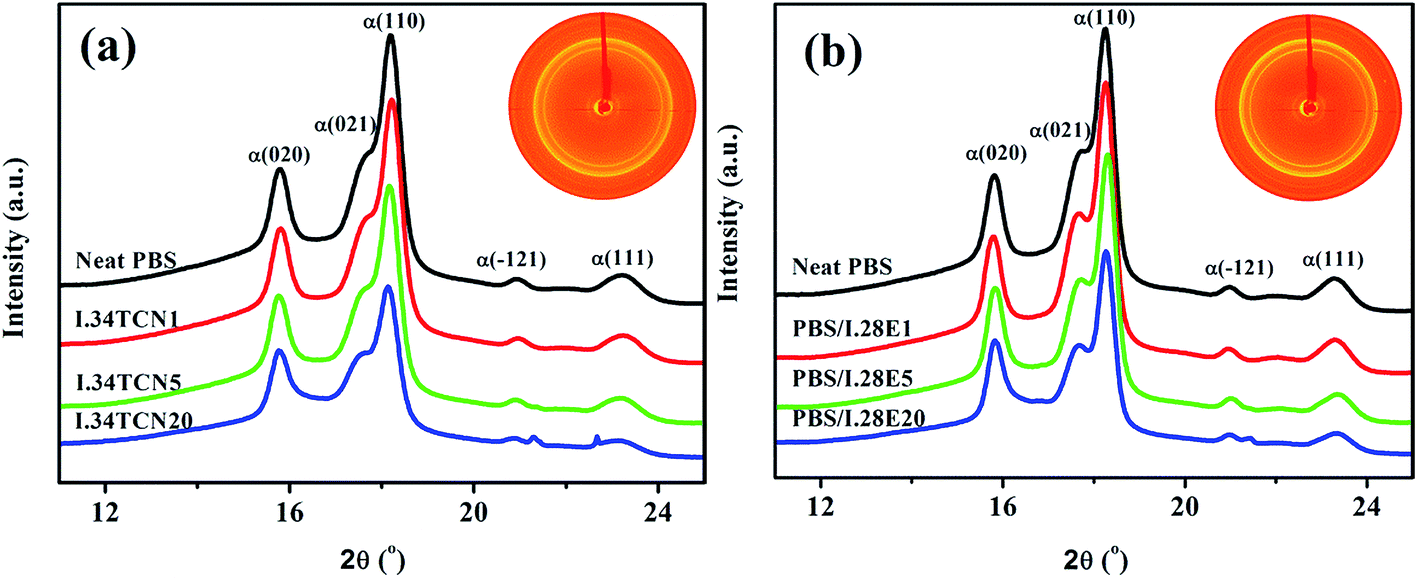 | ||
| Fig. 8 1D-WAXD intensity profiles of PBS/organoclay nanocomposites isothermally crystallized at 100 °C: (a) PBS/I.34TCN nanocomposites, (b) PBS/I.28E nanocomposites. The insets in (a) and (b) are typical 2D-WAXD patterns of PBS/I.34TCN5 and PBS/I.28E5, respectively. | ||
More quantitative information can be acquired from integrated WAXD curves in Fig. 8. And the Xc of PBS/organoclay nanocomposites are calculated based on the 1D-WAXD curves, just as depicted in Fig. 9. It is surprising to find that the Xc value of PBS/I.28E nanocomposites varies from 45–47%, and Xc decreases slightly with the increase of organoclay loading. While in PBS/I.34TCN nanocomposites, the Xc value reduces obviously with the increasing organoclay. For example, the Xc of PBS/I.34TCN20 is 40.6% compared to the neat PBS of 46.9%, the crystallinity drops by 13.4%. Both the confined crystallization of PBS/I.34TCN and PBS/I.28E nanocomposites with the increasing organoclay can be explained by the space restriction of organoclays.18 But the confinement of PBS/I.34TCN crystallization is significantly higher than that of PBS/I.28E at the same condition, probably due to stronger polarity interaction of I.34TCN hinder the diffusion of polymer chains to form nuclei and confine the PBS spherulite for perfection.
 | ||
| Fig. 9 Comparison between PBS/I.34TCN and PBS/I.28E for crystallinity as a function of organoclay contents. | ||
4.4. Understanding the polarity dependence of confined crystallization in the networks of organoclays
It is generally considered that the crystal growth is affected by diffusion and mobility of molecule chains, which is directly related to the crystallization kinetics of polymer. In this works, the crystallization kinetics of PBS was significantly confined by I.34TCN, while enhanced by I.28E at the same mass fraction of organoclay. May the difference between PBS/I.34TCN and PBS/I.28E is only caused by space limitation? Generally, if an organoclay network has been formed or PBS chains sandwiched between organoclay galleries, in which the motions of PBS chains are inhibited, and the confined crystallization behavior should be observed.35–37In order to unambiguously understand the role of I.34TCN and I.28E in PBS for isothermal crystallization, rheological properties of PBS/organoclay nanocomposites are shown in Fig. 10. Fig. 10(a) shows the G′ of neat PBS is monotonically increasing as function of frequency, manifesting a typical shear-thinning flow behavior. Obviously, both G′ of PBS/I.34TCN and PBS/I.28E are sharply increased with the increasing organoclay loading, which demonstrates that the organoclay makes the mobility of PBS chains slow down. At organoclay loading of 20 wt%, both G′ of PBS/I.34TCN20 and PBS/I.28E20 are approximately reached a plateau, independence of the sweeping frequency. The appearance of plateau can be interpreted by formation of organoclay network, which confines diffusion and mobility of PBS chains and shows a solid-like behavior.35 More importantly, it is observed that tanδ (Fig. 10(b)) of PBS/I.34TCN5 is lower than that of PBS/I.28E5 especially in low frequency, indicating that pronounced elastic properties in PBS/I.34TCN5 system and chain mobility will be constrained more obviously, which may be attributed to better compatibility of I.34TCN and PBS compared to I.28E.
 | ||
| Fig. 10 The frequency dependence of (a) storage modulus (G′) and (b) loss tangent (tanδ) for neat PBS, PBS/I.34TCN and PBS/I.28E nanocomposites. | ||
In summary, the crystallization of PBS is complicatedly impacted by organoclay, reinforcing or confining crystallization kinetics. Slightly stronger polarity of I.34TCN decreases the nucleation density, while I.28E acts as heterogeneous nucleating agent. Based on investigation, we propose a comparison of PBS crystallization process in the schematic of Fig. 11 to elaborate the role of surfactant polarity. Both I.34TCN and I.28E are well intercalated and homogeneously dispersed in PBS due to cohesive interaction of organoclay in PBS, both I.34TCN and I.28E are well intercalated and homogeneously dispersed in PBS due to cohesive interaction of organoclay with PBS, a network of organoclay (Fig. 11(a)) could be formed in PBS matrix, which will confine the diffusion and mobility of PBS chains. And stronger interactions lead to confinement effect more obviously, thus, the viscosity of PBS/I.34TCN20 is slightly lower than that of PBS/I.28E20. The thicker constrained layer around I.34TCN (faint yellow area around I.34TCN networks) may form with more vigorous interactions between PBS chains and the modifier of clay due to stronger polarity compared to that of PBS/I.28E (reddish area around I.28E networks). During the stage of nucleation, the nucleation seems to be difficult to occur in constrained layer due to this confinement effect (this stronger polar organic modifier grafted nanoclays act as nucleation inhibitors), and thus the initial nucleation of PBS/I.34TCN under confinement effect may occur far away from this specific region, but initial nucleation can occur in the vicinity of I.28E due to heterogeneous nucleation. Therefore, the nucleation density of PBS/I.34TCN is lower than that of PBS/I.28E (Fig. 11(b)). As organoclay loading increasing, nucleation density is further decreased owing to the spatial constraint.38,39 During the process of crystal growth, PBS spherulites grow rapidly because of templating effect (Fig. 11(c)), its quantity in PBS/I.34CN is less than that in PBS/I.28E, finally, sporadic PBS spherulites are formed in I.34TCN networks and result in low crystallinity of PBS/organoclay nanocomposites.
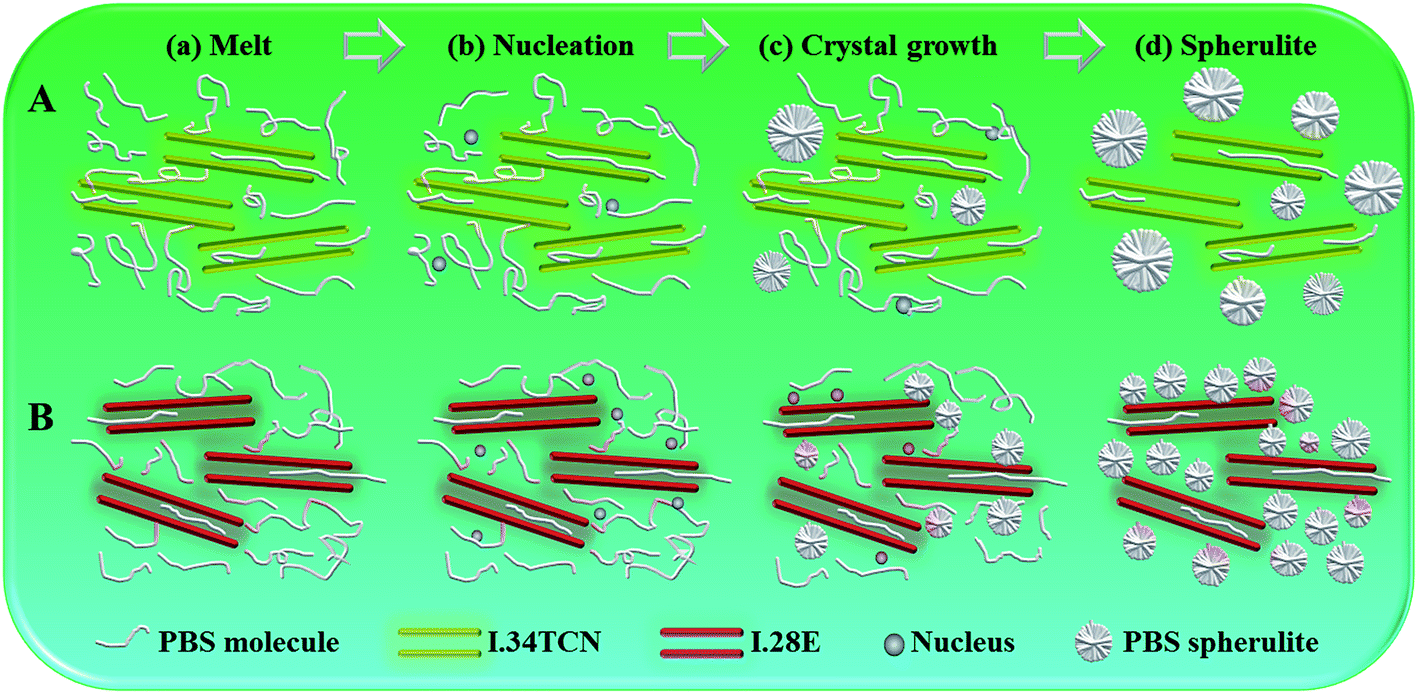 | ||
| Fig. 11 Schematic diagram of comparison of PBS crystallization process in the presence of different types of organoclay: (A) the crystallization process of PBS/I.34TCN, and (B) the crystallization process of PBS/I.28E. | ||
5. Conclusions
We compared and investigated the effect of polarity of organoclay (I.34TCN and I.28E) on the crystallization behavior of PBS. Similar to other organoclay/polymer nanocomposites, two kinds of organically modified clay are well intercalated and homogeneously dispersed in PBS matrix, but it reveals diverse crystallization kinetics. According to the measurement of DSC, all clays facilitates the PBS crystallization kinetics in the case of low loading and suppresses crystallization at higher loading, but the I.34TCN (at low organoclay content) seriously confines the crystallization properties of PBS in comparison to I.28E. All half-crystallization times of PBS intercalated a certain of I.34TCN content are longer than that of PBS/I.28E, manifesting that the I.34TCN retards the PBS crystallization rate. Combining with POM, the nucleation density of PBS/I.34TCN is lower than that of PBS/I.28E, hence, I.34TCN sheets act as a kind of nucleation inhibitor. Moreover, the crystallinity of PBS is significantly decreased compared to neat PBS, while PBS/I.28E blends remain approximately constant. Finally, confined crystallization does not alter the α-form crystal structure of PBS regardless of type of organoclay and its contents. Stronger polar organic modifier grafted nanoclay can significantly retard the formation of nucleation sites, resulting in low crystallization rate and crystallinity.Acknowledgements
The authors gratefully thank the financial support from the National Natural Science of China (Grant No. 21576173, 51273131, 51403139 and 51473101), and State Key Laboratory of Polymer Materials Engineering (sklpme 2014-3-08). Our work was also supported by the Fundamental Research Funds for the Central Universities. The authors are grateful for the kind help and support of Shanghai Synchrotron Radiation Facility (SSRF) in WAXD measurement and the analysis of its results.Notes and references
- A. K. Mohanty, M. Misraa and G. Hinrichsen, Macromol. Mater. Eng., 2000, 276, 1–24 CrossRef.
- T. Ohkita and S.-H. Lee, J. Appl. Polym. Sci., 2005, 97, 1107–1114 CrossRef CAS.
- Y. Someya, T. Nakazato, N. Teramoto and M. Shibata, J. Appl. Polym. Sci., 2004, 91, 1463–1475 CrossRef CAS.
- Y. F. Shih, T. Y. Wang, R. J. Jeng, J. Y. Wu and C. C. Teng, J. Polym. Environ., 2007, 15, 151–158 CrossRef.
- Y. J. Phua, eXPRESS Polym. Lett., 2011, 5, 93–103 CrossRef CAS.
- A. Bhatia, R. K. Gupta, S. N. Bhattacharya and H. J. Choi, J. Nanomater., 2012, 2012, 1–11 CrossRef.
- G.-X. Chen and J.-S. Yoon, J. Polym. Sci., Part B: Polym. Phys., 2005, 43, 817–826 CrossRef CAS.
- S. Y. Hwang, M. J. Ham and S. S. Im, Polym. Degrad. Stab., 2010, 95, 1313–1320 CrossRef CAS.
- V. Ojijo and S. S. Ray, Prog. Mater. Sci., 2014, 62, 1–57 CrossRef CAS.
- S. Pavlidou and C. D. Papaspyrides, Prog. Polym. Sci., 2008, 33, 1119–1198 CrossRef CAS.
- T. D. Fornes and D. R. Paul, Macromolecules, 2004, 37, 7698–7709 CrossRef CAS.
- S. C. Lao, J. H. Koo, T. J. Moon, M. Londa, C. C. Ibeh, G. E. Wissler and L. A. Pilato, J. Fire Sci., 2011, 29, 479–498 CrossRef CAS.
- T. Liu, K. Ping Lim, W. Chauhari Tjiu, K. P. Pramoda and Z.-K. Chen, Polymer, 2003, 44, 3529–3535 CrossRef CAS.
- B. Kim, S.-H. Lee, D. Lee, B. Ha, J. Park and K. Char, Ind. Eng. Chem. Res., 2004, 43, 6082–6089 CrossRef CAS.
- C. I. W. Calcagno, C. M. Mariani, S. R. Teixeira and R. S. Mauler, Polymer, 2007, 48, 966–974 CrossRef CAS.
- V. V. Ginzburg, C. Singh and A. C. Balazs, Macromolecules, 2000, 33, 1089–1099 CrossRef CAS.
- C. S. Triantafillidis, P. C. LeBaron and T. J. Pinnavaia, Chem. Mater., 2002, 14, 4088–4095 CrossRef CAS.
- J.-B. Chen, J.-Z. Xu, H. Xu, Z.-M. Li, G.-J. Zhong and J. Lei, Chin. J. Polym. Sci., 2015, 33, 576–586 CrossRef CAS.
- S.-Y. Zhou, J.-B. Chen, X.-J. Li, X. Ji, G.-J. Zhong and Z.-M. Li, RSC Adv., 2016, 6, 2530–2536 RSC.
- D. Xu and Z. Wang, Polymer, 2008, 49, 330–338 CrossRef CAS.
- Z. Liu, K. Chen and D. Yan, Eur. Polym. J., 2003, 39, 2359–2366 CrossRef CAS.
- D. S. Homminga, B. Goderis, V. B. F. Mathot and G. Groeninckx, Polymer, 2006, 47, 1630–1639 CrossRef CAS.
- D. Homminga, B. Goderis, I. Dolbnya, H. Reynaers and G. Groeninckx, Polymer, 2005, 46, 11359–11365 CrossRef CAS.
- V. Miri, S. Elkoun, F. Peurton, C. Vanmansart, J.-M. Lefebvre, P. Krawczak and R. Seguela, Macromolecules, 2008, 41, 9234–9244 CrossRef CAS.
- S. D. Benson and R. B. Moore, Polymer, 2010, 51, 5462–5472 CrossRef CAS.
- T. D. Fornes and D. R. Paul, Polymer, 2003, 44, 3945–3961 CrossRef CAS.
- E. Di Maio, S. Iannace, L. Sorrentino and L. Nicolais, Polymer, 2004, 45, 8893–8900 CrossRef CAS.
- J. Shi, X. Yang, X. Wang and L. Lu, Polym. Test., 2010, 29, 596–602 CrossRef CAS.
- Z. Lv, K. Wang, Z. Qiao and W. Wang, Mater. Des., 2010, 31, 3804–3809 CrossRef CAS.
- A. R. Morales, L. B. d. Paiva, D. Zattarelli and T. R. Guimarães, Polimeros, 2012, 22, 54–60 CAS.
- J. Y. Nam, S. S. Ray and M. Okamoto, Macromolecules, 2003, 36, 7126–7131 CrossRef CAS.
- C. J. Perez and V. A. Alvarez, J. Appl. Polym. Sci., 2009, 114, 3248–3260 CrossRef CAS.
- M. Avella, S. Cosco, G. D. Volpe and M. E. Errico, Adv. Polym. Technol., 2005, 24, 132–144 CrossRef CAS.
- M. Kennedy, G. Turturro, G. R. Brown and L. E. St-Pierre, Nature, 1980, 287, 316–317 CrossRef CAS.
- X.-F. Wei, R.-Y. Bao, Z.-Q. Cao, W. Yang, B.-H. Xie and M.-B. Yang, Macromolecules, 2014, 47, 1439–1448 CrossRef CAS.
- T. Wang, H. Li, F. Wang, S. Yan and J. M. Schultz, J. Phys. Chem. B, 2011, 115, 7814–7822 CrossRef CAS PubMed.
- T. Wang, H. Li, F. Wang, J. M. Schultz and S. Yan, Polym. Chem., 2011, 2, 1688 RSC.
- D. Homminga, B. Goderis, I. Dolbnya and G. Groeninckx, Polymer, 2006, 47, 1620–1629 CrossRef CAS.
- K.-h. Nitta, K. Asuka, B. Liu and M. Terano, Polymer, 2006, 47, 6457–6463 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2016 |