
Photo-doping of plasma-deposited polyaniline (PAni)†
 ac
ac
Abstract
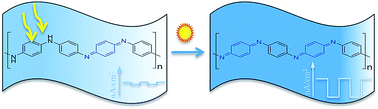
Although polyaniline (PAni) has been studied extensively in the past, little work has been done on producing films of this material via plasma deposition. We have synthesized and analysed the photoresponse behavior of plasma-deposited polyaniline films and proceeded to dope the films using light and with various metal ions. Upon illumination, the photocurrent responses of the thin plasma films increased over time, and the response was dependent on the film thickness. On doping the film with metal ions, the photocurrent densities were enhanced from nano- to micro-amperes per square centimeters. Doping seemed, however, to cause the films to become unstable. Despite this setback, which requires further research, the drastic increase in current shows great promise for the development of plasma-deposited polyaniline films for application in the area of organic electronics and photovoltaics.
 Please wait while we load your content...
Please wait while we load your content...

