
High yield synthesis of amine functionalized graphene oxide and its surface properties†
Souvik Chakrabortya,
Saikat Sahaa,
V. R. Dhanakb,
Kallolmay Biswasc,
Michel Barbezatd,
Giovanni P. Terrasid and
Amit K. Chakraborty*a
aCarbon Nanotechnology Lab, Department of Physics, National Institute of Technology Durgapur, Mahatma Gandhi Avenue, Durgapur – 713209, West Bengal, India. E-mail: amitkc61@gmail.com
bDepartment of Physics and Stephenson Institute for Renewable Energy, University of Liverpool, Liverpool, L69 3BX, UK
cJubilant Chemsys Ltd., Sector 58, B-34, Noida – 201301, Uttar Pradesh, India
dLaboratory for Mechanical Systems Engineering, Empa, Swiss Federal Laboratories for Materials Science & Technology, Überlandstrasse 129, 8600 Dübendorf, Switzerland
First published on 11th July 2016
Abstract
Graphene and its derivatives have attracted great research interest due to their many exciting properties leading to a number of potential applications. However, for many practical applications including reinforcement in epoxy resin matrices, chemical functionalization of graphene is often a necessary requirement. Herein we report a simple temperature-assisted reflux method to synthesize graphene oxide (GO) functionalized with n-butylamine in high yield without the use of toxic chemicals. X-ray photoelectron spectroscopy and energy dispersive analysis by X-rays showed successful attachment of amine groups onto GO to form (GO-ButA). The functionalized GO was further characterized using Raman, nuclear magnetic resonance and Fourier transform infrared spectroscopies. The surface morphology and particle size, etc. were characterized using scanning electron microscopy. The solution properties of the GO-ButA were investigated by dispersing in water and common organic solvents which indicated an increased hydrophobic nature of the product which was also confirmed by contact angle measurements. Further, the interaction of GO-ButA with epoxy resin was tested by dispersing it in an epoxy resin which indicated an improved and more stable dispersion as compared to that of GO which shows the potential application of GO-ButA as a reinforcement (filler) in epoxy based composites.
1. Introduction
Since its successful isolation by Novoselov et al.,1 graphene has become a very important member of the nanomaterials family exhibiting interesting electrical, mechanical and thermal properties suitable for a wide range of applications from sensors to supercapacitors to reinforcing fillers in structural polymer composites.2–8 However, difficulty in large scale synthesis combined with relative inertness due to lack of functional surface groups often limit its full potential in structural composite applications.9 Recent advances in the bulk production10–13 of graphene and its derivatives such as graphene oxide (GO), reduced graphene oxide (rGO), functionalized graphene nanosheets (FGS) etc. have overcome many of the limitations that have hindered graphene's application. One of several techniques reported for the large scale synthesis of graphene is chemical reduction of its oxide (GO)13 which is produced primarily by strong acid treatment of natural graphite as reported by Hummers and Offeman.14 This method allows for large scale synthesis of single to few layered graphene oxides suitable for applications requiring gram quantities of graphene such as in structural polymer composites where GO plays the role of the filler.For application in polymer based structural composites, often the preferred matrix material is epoxy resins due to their good thermal, chemical and mechanical stability. Hence, studies on improving the properties of epoxy matrix by incorporation of nanoscale fillers such as carbon nanotubes (CNT), graphene, and ceramic nanoparticles have received great interest in the recent past.15–19 Generally, the effective reinforcement of polymer matrices is largely dependent on two criteria: (i) uniform and stable dispersion of the filler in the matrix and (ii) better interfacial interaction between the filler and the matrix. But the use of pristine graphene as filler in epoxy matrices has been rather limited mainly due to its inability to achieve uniform and stable dispersion.20 Studies on CNT, the one dimensional cylindrical allotrope of graphene, have shown that chemical functionalization often improves their solution properties21–24 and the same can be said for graphene.25 Thermally and chemically modified graphene oxides have been used to reinforce epoxy matrices with significant success in improving their mechanical properties.20,26–31
One of the most common methods of chemical functionalization employed for this purpose is attachment of amino groups to the filler surfaces which apparently improves the filler–epoxy matrix interaction leading to an improvement in the mechanical properties of the cured composites.30,32–37 This is generally explained by a possible ring opening nucleophilic substitution reaction by the –NH2 moiety attacking the epoxy group in the resin which leads to a better filler matrix interaction.32,34 Amino functionalization of nanofillers is generally carried out by acylation38 which requires the use of toxic chemicals like thionyl/acyl chloride which not only raises safety concerns for handling but also pollution concerns for environment and hence is not desirable. An alternative method in which an alcoholic solution of an amine precursor is used for amination at room temperature39 avoids use of toxic chemicals but here the yield is quite low. In particular, for short chain aliphatic amines, such as n-butylamine, we found a yield of less than 100 mg product per gm of reactants using this method.
For applications in epoxy based composites, one requires sufficient quantities of functionalized filler at a reasonable cost and hence currently, there is a large demand from the industries for functionalization methods that offer high yield synthesis at relatively low cost and are environment friendly. Li et al.40 reported a temperature assisted reflux method to synthesize octadecylamine (long-chain) functionalized GO (without using acylation) resulting in reduction of GO with highly hydrophobic surfaces. However, the use of such fillers in epoxy based composites is rather limited as the long alkyl chains may cause significant softening influence to the mechanical properties of the composite. Therefore, at present, there is a growing demand for environment-friendly methods which allow for high-yield synthesis of GO functionalized with short-chain amines for application in epoxy based composites as short alkyl chains will have limited softening influence but they can offer sufficient steric hindrance to discourage filler re-agglomeration in the cured composites. In view of the above, here we report, for the first time, a facile eco-friendly method (avoiding the use of thionyl/acyl chloride) for amination of GO with n-butyl amine (a short-chain amine) that gives high yields of (∼30%) aminated GO (GO-ButA).
The success of the method has been confirmed by X-ray photoelectron spectroscopy (XPS), Fourier transform infrared (FTIR), Raman and energy dispersive X-ray (EDX) spectroscopic studies. The surface morphology of both GO and GO-ButA has been investigated via SEM. The solution property of the GO-ButA has been investigated by testing its dispersion in different common organic solvents and water. Contact angle measurements have been carried out to probe the hydrophobicity of the functionalized material. Further, the effect of this functionalization on the dispersion of GO-ButA (e.g., stability and uniformity) in an epoxy resin has been systematically investigated with a goal to evaluate the potential of the functionalized GO as reinforcing filler in the epoxy matrix as we know that a uniform and stable dispersion of the filler is the first step to effective reinforcement of a composite.
2. Experimental
2.1 Materials
Natural graphite flakes were procured from Kai Yu Industries, Nanjing, China. NaNO3 (≥99.5%), KMnO4 (min 99%) and H2SO4 (95–98%) were procured from Merck, India and n-butylamine (99.5%) from Sigma Aldrich. Ethanol (min 99.9%) was purchased from Changshu Yangyuan Chemicals, China. The bisphenol-A based epoxy resin – LAPOX L-12 (Atul Co., India) was procured from The Perfect Trading Company, Kolkata, India. All ingredients and chemicals were used as purchased without further purification.2.2 Preparation of graphene oxide (GO)
GO was prepared following the Hummers'14 method. Natural graphite flakes (3 g) were first added to a beaker containing conc. H2SO4 (98 mL, 95–98%) placed in an ice bath in order to maintain a temperature below 10 °C. The ice bath was placed on a magnetic stirrer so that the flakes inside the beaker are stirred. In the next step, NaNO3 (1.5 g) was added in batches to the reaction mixture followed by the addition of KMnO4 (9 g) while keeping the temperature below 18 °C for 30 min. Subsequently the temperature was allowed to rise to 35 °C and the stirring continued for further 30 min. Next, distilled water (130 mL) was added dropwise while ensuring that the temperature did not rise above ∼98 °C. 20 mL of H2O2 (30%) was added to the mixture and then allowed to stand till a clear supernatant was obtained. This supernatant was then decanted and the remaining reaction mixture was diluted with distilled water. This process was repeated till the pH became neutral. The solution thus obtained was centrifuged to get a brownish paste which was dried overnight under vacuum at 80 °C to get GO.2.3 Preparation of n-butylamine functionalized graphene oxide (GO-ButA)
GO (300 mg) was dispersed in distilled water (150 mL) in a glass beaker using an ultrasonic probe sonicator (BioMatrix, India, 500 W) operating at 40% amplitude for 10 min with an ON/OFF pulse of 5 s/10 s. In a separate beaker, n-butylamine (450 mg) was dispersed in ethanol (45 mL, 99% pure) via bath sonication for 10 min. The GO suspension was then transferred to a 500 mL round bottom flask in which the n-butylamine suspension was also transferred and the mixture stirred in an oil bath under reflux condition at 60 °C for 20 h. The black solution eventually obtained was left to stand undisturbed for 24 h after which it showed a separation of the functionalized graphene oxide from the solvent suggesting its hydrophobic nature. The functionalized graphene oxide was then extracted via solvent evaporation under vacuum. The obtained solid product was washed several times with ethanol via bath-sonication before extracting the black powdery GO-ButA by overnight solvent evaporation under vacuum at 80 °C. The average yield per batch was found to be around 250 mg (the total mass of the reactants was 750 mg).2.4 Characterization
XPS measurements were performed using a Scienta SES-200 analyzer operated in ultra-high vacuum using MgKα X-ray source emitting photons of energy 1253.6 eV. A Shimadzu IRAffinity 1S system with diamond ATR analyzer was used for the FTIR analysis. A Carl Zeiss ΣIGMA field emission scanning electron microscope operating at an accelerating voltage of 5 kV was used to capture SEM micrographs. Average values of the particle size were estimated by the Gaussian fits of the histograms plotted by measuring the sizes of the particles using ImageJ software (total 75 particles from three SEM micrographs). EDX spectra were obtained using an X-MAX 50 detector from Oxford Instruments, UK which was attached to the SEM chamber. 1H NMR spectra were recorded on a Bruker Ultrashield™ (400 MHz) instrument and calibrated to residual solvent peak (DMSO-D6 at 2.50 ppm). Raman analysis was carried out using a Horiba Jobin Yvon T64000 Raman triple spectrometer using a Spectra Physics Stabilite 2017 argon ion laser of wavelength 514.5 nm as the excitation source. TGA of the as-synthesized GO and GO-ButA were carried out in a Perkin Elmer Simultaneous Thermal Analyzer, STA 6000 in an uncapped alumina crucible operated at a heating rate of 10 °C min−1 in nitrogen atmosphere. Contact angle made by a water droplet (∼5 μl in volume) with the surface of a pellet (dia ∼ 13 mm, thickness ∼ 1 mm) made of the sample was measured using a Dataphysics OCA20 instrument. Optical microscopy analysis was carried out with an Olympus BX51 polarizing optical microscope under transmission mode using unpolarized light. The micrographs were captured using an Olympus DP12 camera with the help of AnalySIS software.Sampling for optical microscopy analysis was carried out by drop casting a fixed mass of the epoxy dispersion (∼0.014 g) onto a clean glass slide. The slide was then covered with another slide and they were both pressed under a weight (∼2.25 kg) for 10 min. The above steps were followed to get an almost uniform film thickness (∼0.02 mm) for each of the analyzed samples. A fresh sample was prepared each time for microscopy analysis from the original dispersion which had been stored at room temperature.
3. Results and discussion
3.1 XPS analysis
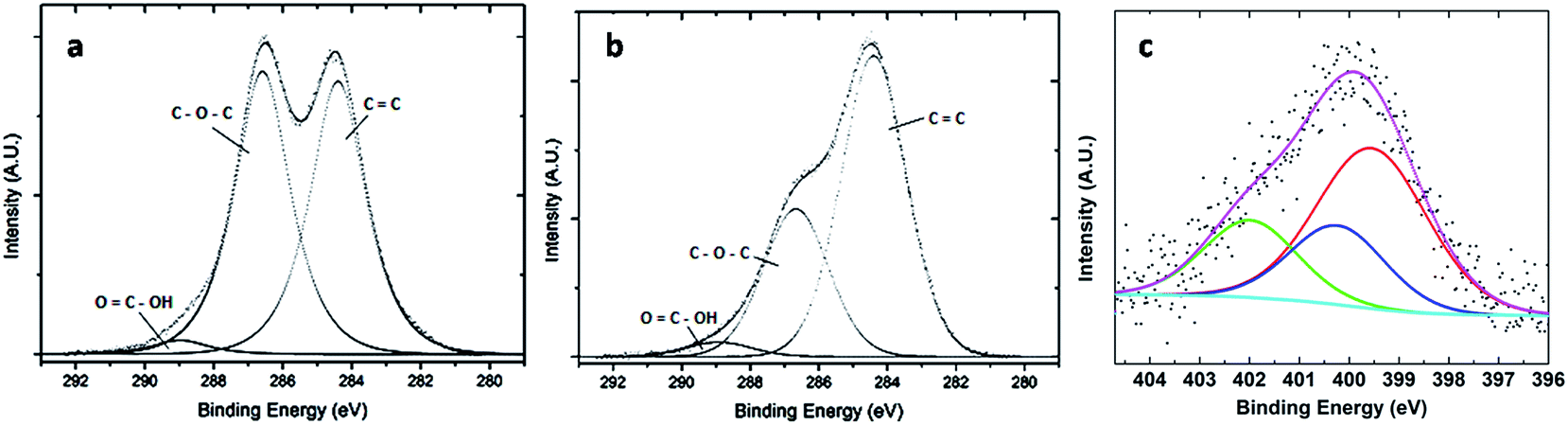
Fig. 1a and b show the photoelectron spectra of C1s electrons for GO and GO-ButA, respectively. The spectra were fitted with mixed Gaussian–Lorentzian function and deconvoluted into components using the CASAXPS program. Fig. 1a shows two clearly resolved carbon 1s peaks: one at binding energy of 284.4 eV due to sp2 bound graphitic carbon (indicated by C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond in the figure) and the other at binding energy of 286.6 eV due to carbon atoms attached to oxygen in the epoxide (C–O–C) groups with almost equal intensity. A small peak at a binding energy of about 288.9 eV is also observed which corresponds to carboxylic acid groups (–COOH) attached to the edges of GO.43,44 Fig. 1b shows similar carbon 1s spectrum except that the intensity of the peak at 286.6 eV is significantly reduced which is consistent with the successful reduction of GO upon n-butylamine treatment.45 This is a remarkable finding considering that most chemical methods for synthesis of graphene requires chemical reduction by hydrazine to form reduced GO in which evolution of N2 gas is known to occur during reduction which may lead to local heterogeneities in the graphene surface.44 Further, the removal of oxygen containing functionalities by hydrazine allows for tighter packing of the sheets which is not desirable for filler application where one requires the surface area to be the highest in order for maximum interaction with the matrix. By eliminating the need for a separate reduction step, our method ensures that the graphene surfaces are not only well-separated from each other even after reduction due to long alkyl chains acting as spacer between the sheets but also less defective (as gases evolved, if any will diffuse easily through large inter-sheet gap).
C bond in the figure) and the other at binding energy of 286.6 eV due to carbon atoms attached to oxygen in the epoxide (C–O–C) groups with almost equal intensity. A small peak at a binding energy of about 288.9 eV is also observed which corresponds to carboxylic acid groups (–COOH) attached to the edges of GO.43,44 Fig. 1b shows similar carbon 1s spectrum except that the intensity of the peak at 286.6 eV is significantly reduced which is consistent with the successful reduction of GO upon n-butylamine treatment.45 This is a remarkable finding considering that most chemical methods for synthesis of graphene requires chemical reduction by hydrazine to form reduced GO in which evolution of N2 gas is known to occur during reduction which may lead to local heterogeneities in the graphene surface.44 Further, the removal of oxygen containing functionalities by hydrazine allows for tighter packing of the sheets which is not desirable for filler application where one requires the surface area to be the highest in order for maximum interaction with the matrix. By eliminating the need for a separate reduction step, our method ensures that the graphene surfaces are not only well-separated from each other even after reduction due to long alkyl chains acting as spacer between the sheets but also less defective (as gases evolved, if any will diffuse easily through large inter-sheet gap).
 | ||
| Fig. 1 XPS spectra (experimental and the fitted curves with deconvoluted components). (a) C1s peak of GO, (b) C1s peak of GO-ButA and (c) N1s peak of GO-ButA. | ||
Fig. 1c shows the N1s spectrum obtained from the GO-ButA sample in which a broad peak at binding energy of 400.2 eV is observed which upon deconvolution shows presence of one main component (50%) at binding energy of 399.6 eV and two other smaller components at energies of 400.3 eV (23%), and 401.9 eV (23%). The component peaking at 399.6 eV indicates the formation of C–N bonds with secondary amines (C–NH–R) whereas the other two are due to presence of amide (C(O)–NH–R) groups, and alkylammonium ions (R–NH3+), respectively, in accordance with previous literature,44 where R represents butyl group.
Based on the above findings, a schematic model of the chemical structures which is consistent with our data for the synthesis of GO-ButA is presented in Fig. 2. The main interaction between the amines and graphene oxide is via ring-opening aminations of epoxides to yield 1,2-amino alcohols which accounts for 54% of the nitrogen. In addition, n-butylamine interacts with carboxylic acid groups present along the sheet edges to form secondary amides. Evidence of amide formation as well as alkyl chain attachment is also observed from the NMR analysis (details in Fig. S1 of ESI†).
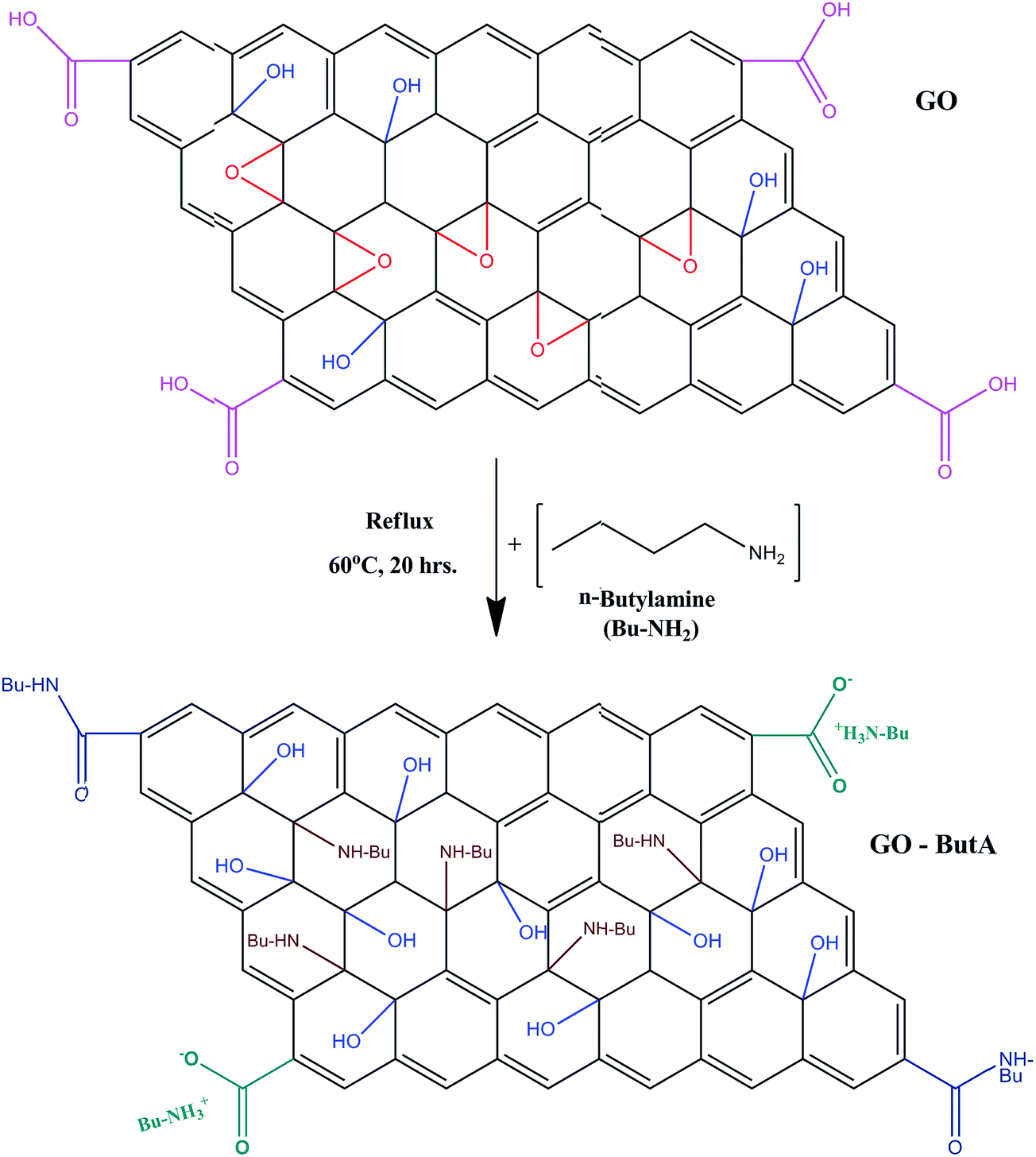 | ||
| Fig. 2 A simple chemical model representing the formation of GO-ButA from GO. | ||
Quantitative information on the relative atomic concentrations of the three main elements present in the samples as obtained from the analysis is summarized in Table 1. In the table, the relative contribution of each of the component of the carbon peak calculated with respect to the total carbon concentration is also shown. It is evident that the graphitic carbon fraction has increased by about 21 wt% upon functionalization of GO. The C1s component due to carbon bound to oxygen atoms in epoxide groups also show a reduction of almost same percentage (24 wt%) upon functionalization. This is very much consistent with our proposed model of amine attachment via opening of the epoxide rings.
| Sample | Relative weight (at%) | |||||
|---|---|---|---|---|---|---|
| Components of carbon in atomic weight fraction (% C) | Carbon | Oxygen | Nitrogen | |||
| CC |
C–O–C | OC–OH |
||||
| GO | 47.9 | 49.7 | 2.4 | 62.7 | 37.3 | 0 |
| GO-ButA | 68.7 | 25.8 | 5.5 | 66.7 | 28.0 | 5.3 |
3.2 FTIR analysis
As is seen from the Fig. 3, the FTIR spectra of GO is characterized by the presence of peaks for C–O bond stretching of the epoxy group in the range 970 to 1150 cm−1, C–OH bond stretch at around ∼1370 cm−1 and CO stretching for carbonyl and carboxyl groups at around ∼1715 cm−1 and ∼1615 cm−1 respectively.40,46,47 The broad peak around ∼3200–3500 cm−1 indicates the presence of –OH group (H bonded) in GO.46 Upon functionalization (red curve in Fig. 3), two new peaks ∼2924 cm−1 and ∼2958 cm−1 appear due to C–H stretching vibrations of the aliphatic chains of the amines attached onto the GO surface.40 These peaks which appear only in the presence of alkyl chains validate our claim for the successful synthesis of GO-ButA. Disappearance of the peak at ∼1715 cm−1 and a significant decrement in peak intensity within ∼3200–3500 cm−1 show signs of reduction of GO upon functionalization. The spectrum for GO-ButA also shows a weak and somewhat broad peak at ∼3660 cm−1 and a strong peak at ∼1580 cm−1 which are associated with the N–H stretching vibrations of the secondary amines and the CO stretching of secondary amides, respectively which provides further evidence of the formation of amine and amide bonds in GO-ButA37 consistent with our XPS data in Fig. 1c.
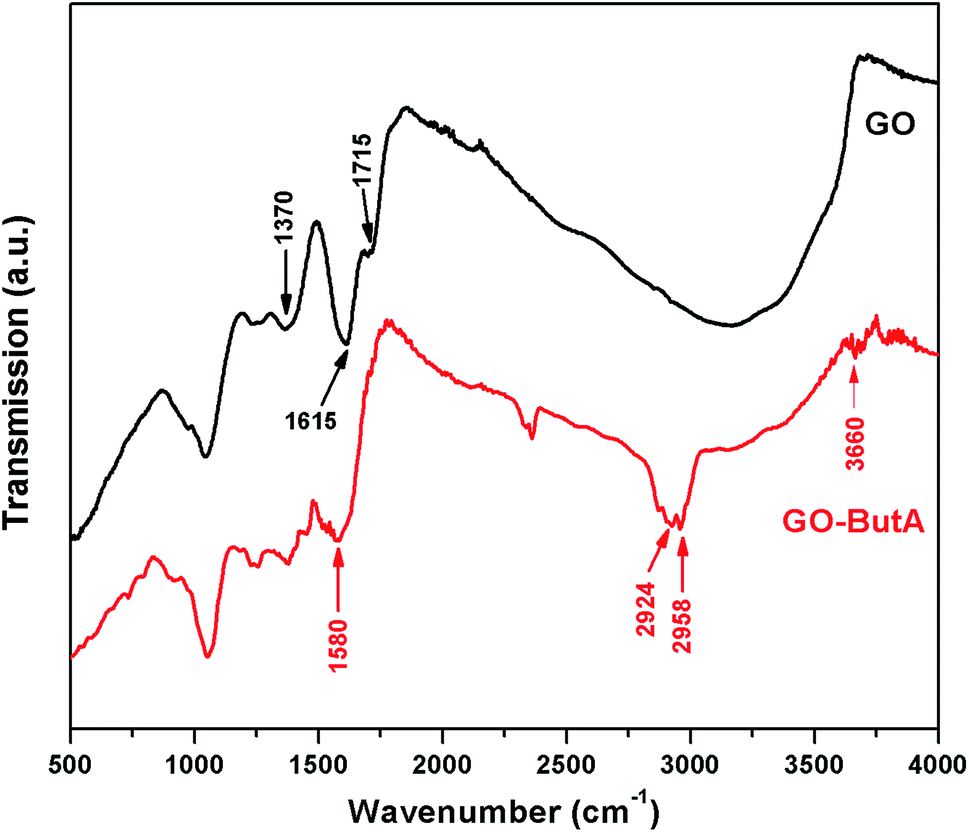 | ||
| Fig. 3 FTIR spectra of the GO and GO-ButA. | ||
3.3 Raman analysis
For carbons, the main attributes for a Raman spectra is characterized by the D and G peaks present around 1360 cm−1 and 1580 cm−1 for excitations within the visible range.48 The D peak, generated by the zone boundary phonons, provides information about the defects and impurities present in it and the G peak arises due to the doubly degenerate zone centre E2g mode and is omnipresent for all carbon based materials.48,49 Upon oxidizing graphite – to derive GO, we see (Fig. 4) a shift in the position of the G-peak (at 1598 cm−1) of the Raman spectrum to higher wave number. Fig. 4 also shows that the position of the G-peak is further shifted to 1593 cm−1 for GO-ButA. This is consistent with the observation of Li et al.40 in which the G-peak was found to shift from 1596 cm−1 to 1588 cm−1 upon functionalization with octadecylamine which the authors attributed to the restoration of sp2 graphitic network. The intensity ratio IG/ID for both GO and GO-ButA are found to be around 1.02 indicating similar degrees of disorder which is understandable considering that functionalization of GO occurs by attachment of amines predominantly on the defect sites where oxidizing groups were already present and hence it does not generally involve conversion of sp2 bonds into sp3.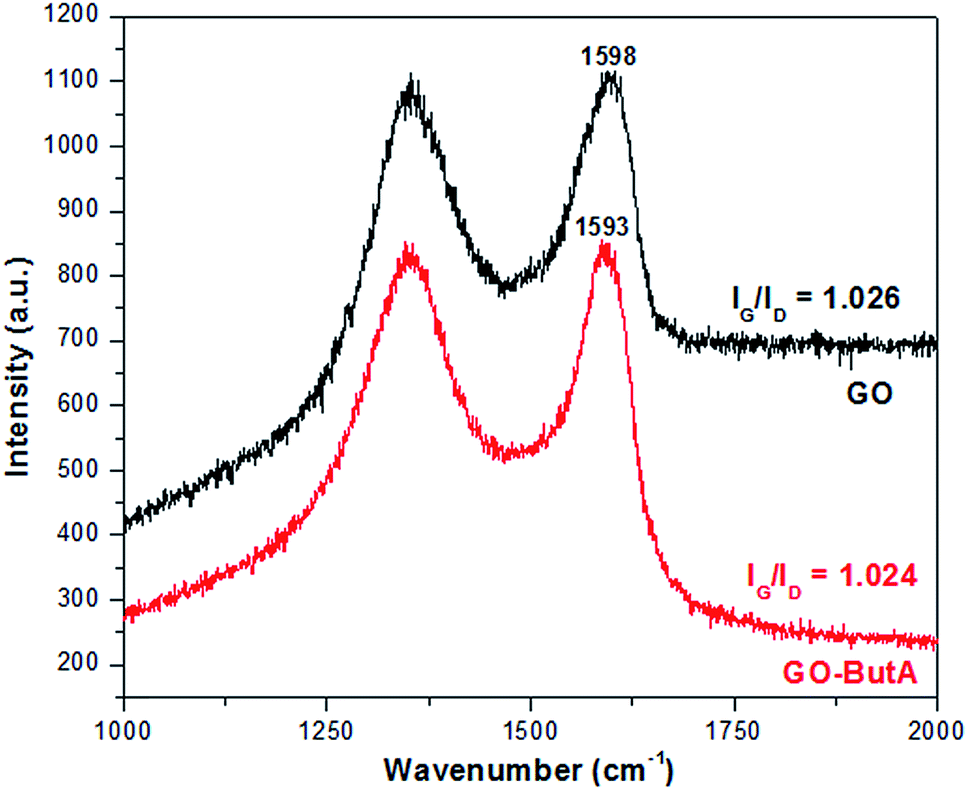 | ||
| Fig. 4 Raman spectra of GO and GO-ButA. Notice the shift in the G peak of the spectra towards that of pristine graphite (1580 cm−1) upon functionalization. | ||
3.4 SEM and EDX analysis
The SEM micrograph in Fig. 5a and b reveal rough surfaces with extensive presence of ripple-like structure on GO whereas Fig. 5c shows the histogram plot of the particle (flake) size distribution showing an average flake size of ∼8 μm. Fig. 5d and e depict the micrographs obtained from GO-ButA, which appear to be smoother compared to those of GO. The increased smoothness of the surface may be due to increased spacing between the sheets due to incorporation of alkyl chains onto the plane of the sheets which allows for easy release/diffusion of any gases evolved during functionalization thus preventing defects to be formed on the surface. The average flake length as calculated from the histogram of GO-ButA (Fig. 5f) is found to be ∼5 μm which is much smaller compared to that of GO (∼8 μm) possibly due to improved exfoliation of the sheets upon functionalization.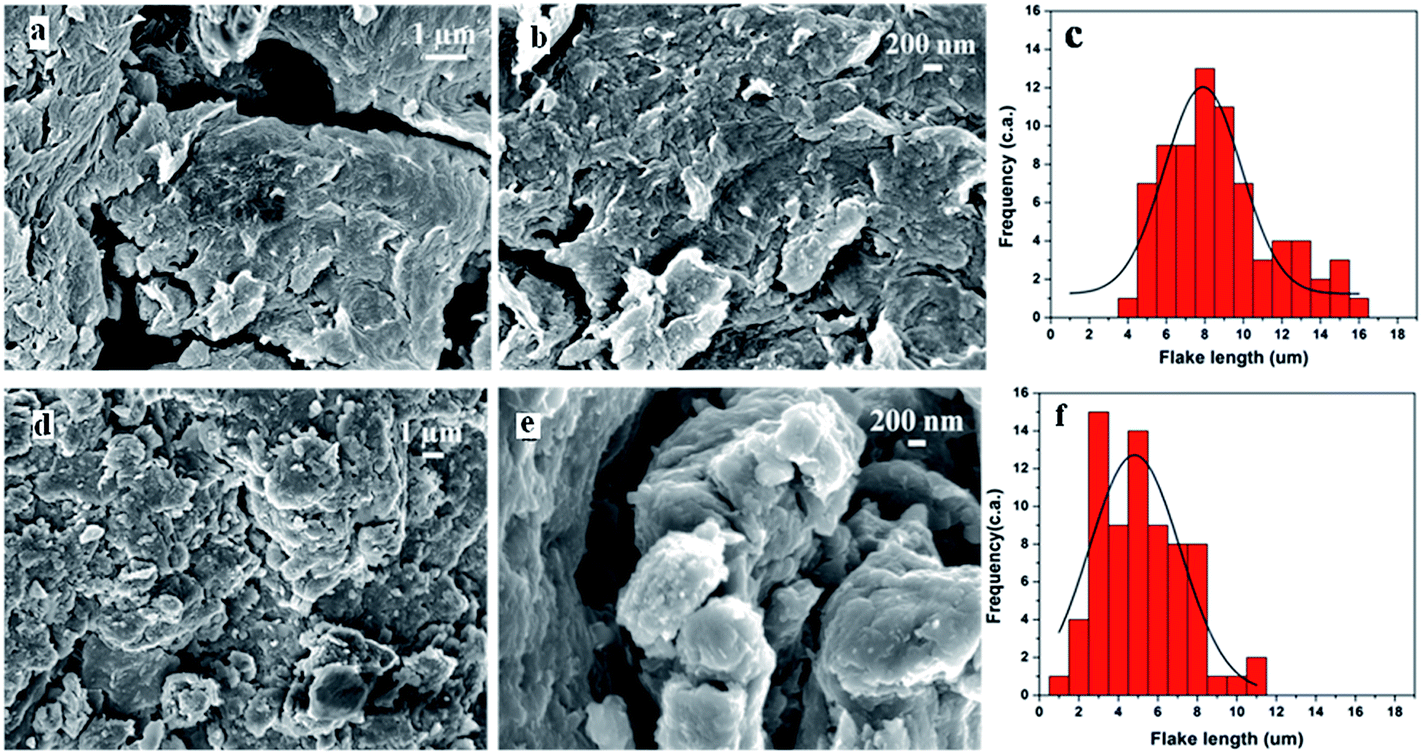 | ||
| Fig. 5 SEM images of GO (a and b) and GO-ButA (d and e). Histogram plots of flake size distribution as obtained from images of GO (c) and GO-ButA (f). | ||
The results in Table 2 are in good agreement with those obtained by XPS analysis as shown in Table 1. The oxygen fraction decreases by about 16 at% from 42 at% in GO to 26 at% in GO-ButA consistent with XPS data in which the reduction was about 10 at%. Both EDX and XPS data show an increase in the carbon fraction by about 6 at% upon functionalization. The differences observed in the estimate of the elements in Tables 1 and 2 may be linked with the sensitivity of detection of the two methods – for example, XPS is a surface sensitive method compared to EDX and hence may not represent the bulk distribution accurately.
| Sample | Atomic fraction of elements (%) | ||
|---|---|---|---|
| Carbon | Oxygen | Nitrogen | |
| GO | 57.7 | 42.3 | 0 |
| GO-ButA | 64.4 | 26.1 | 9.5 |
3.5 Dispersion studies
 | ||
| Fig. 6 Photographs of vial containing dispersion of GO and GO-ButA in various solvents. (a) Taken just after sonication and (b) taken after two weeks from sonication. | ||
 | ||
| Fig. 7 Contact angle for GO-ButA at room temperature and after 30 min annealing at 120 °C. | ||
 | ||
| Fig. 8 Optical micrographs of dispersions of GO (a and b) and GO-ButA (c and d) just after sonication. Filler fraction fixed at 0.2% w/w and magnification 100×. | ||
With GO-ButA as filler, the particulate size is observed to decrease consistently with time (Fig. 9d–f) and the size is smallest for the micrograph recorded 120 min after sonication. On the other hand, GO shows an increase in particulate size within the same time frame (Fig. 9a–c) with some bigger sized particulates (average of 10–30 μm in size) seen scattered amongst the fine particulates (encircled white in Fig. 9b and c) as well. These observations could be interpreted as improved dispersion stability of GO-ButA in epoxy resin compared to that of GO possibly due to reaction of the amine/amide groups of GO-ButA with abundant epoxy groups present in the resin to form ternary amines and amides.34,52
 | ||
| Fig. 9 Optical micrographs after sonication. (a) and d) After 30 min, (b) and (e) after 90 min and (c) and (f) after 120 min. Filler fraction fixed at 0.2% w/w and magnification 100×. | ||
Interestingly, for GO, the particulates observed in the optical micrographs taken just after dispersion into epoxy (Fig. 10a) disappear after seven (Fig. 10b) and fourteen days (Fig. 10c) from sonication. However, micrographs with GO-ButA as fillers still showed particulate evidence (Fig. 10 e and f). An inspection during disposing off the dispersions after fifteen days, revealed that a considerable amount of GO had sedimented at the bottom of the container as a thick opaque layer (Fig. 10d) whereas only a thin layer sedimentation had occurred for GO-ButA (Fig. 10g). This is perhaps, again an indication that the dispersion of GO-ButA is more stable in epoxy matrix than GO (possibly due to its improved interaction with epoxy) which in turn indicates that composites made with GO-ButA as filler are likely to show improved mechanical properties than those made with GO.41
 | ||
| Fig. 10 Optical micrographs of GO and GO-ButA, respectively in epoxy resin: (a) and (e) just after sonication, (b) and (f) after 7 days, (c) and (g) after 14 days from sonication and (d) and (h) photograph of precipitate (indicated by the white arrow) observed at the bottom of the beaker after disposing the supernatant. All micrographs are of magnification 100×. | ||
4. Conclusion
A facile one-step method for high yield synthesis of functionalized graphene (GO-ButA) with short chain alkyl amine has been reported without the use of toxic chemicals such as thionyl/acyl chlorides. The method also eliminates the use of a separate reduction step which is often a necessary step for chemical synthesis of reduced GO. The attachment of amine functionalities to the GO nanosheets has been confirmed by EDX analysis by the presence of nitrogen. The N1s XPS spectrum further confirmed that the nitrogen is indeed in the form of secondary amine and amide based on which we have proposed a model for the reaction mechanism. XPS and EDX analysis also confirmed the successful reduction of GO during this method as the oxygen signal significantly reduced upon functionalization. Evidence of functionalization has also been found from FTIR, NMR and Raman spectroscopic analysis. The surface morphology of the GO-ButA has been evaluated by SEM analysis. That GO-ButA has an improved interaction with an epoxy resin providing better dispersion stability has also been demonstrated. Optical microscopy studies using GO-ButA as filler in an epoxy matrix further revealed that the optimum dispersion condition with minimum agglomerate size is achieved at 120 min after sonication. As many of the common epoxy resins require curing for couple of hours, the functionalized GO produced in this work may find its use as filler in epoxy based composites.Acknowledgements
AKC is grateful to Empa for sponsoring the project in the form of a studentship to SC to carry out this research. The authors acknowledge Dr Subhratanu Bhattacharya and DST-FIST facilities of Department of Physics, Kalyani University for the FTIR measurements. The authors are also thankful to Mrs Anja Huch of Empa for carrying out the contact angle measurements. AKC acknowledges partial financial support of the MHRD, Govt. of India for the sanction of the Centre of Excellence in Advanced Materials (grant ref. No. AC/MHRD/TEQIP-II/CoE/2013, dated 4th April, 2013).References
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Fisorv, Science, 2004, 306, 666 CrossRef CAS PubMed
.
- S. Gupta Chatterjee, S. Chatterjee, A. K. Ray and A. K. Chakraborty, Sens. Actuators, B, 2015, 221, 1170 CrossRef CAS
- N. Chakrabarty, S. Sivaprakash and A. K. Chakraborty, AIP Conf. Proc., 2015, 1665, 050072 CrossRef
- W. Li, A. Dichiara and J. Bai, Compos. Sci. Technol., 2013, 74, 221 CrossRef CAS
- Y. Wang, Q. He, H. Qu, X. Zhang, J. Guo, J. Zhu, G. Zhao, H. A. Colorado, J. Yu, L. Sun, S. Bhana, M. A. Khan, X. Huang, D. P. Young, H. Wang, X. Wang, S. Wei and Z. Guo, J. Mater. Chem. C, 2014, 2, 9478 RSC
- Y. Wang, H. Wei, Y. Lu, S. Wei, E. K. Wujcik and Z. Guo, Nanomaterials, 2015, 5, 755 CrossRef CAS
- H. Yi, H. Wang, Y. Jing, T. Peng, Y. Wang, J. Guo, Q. He, Z. Guo and X. Wang, J. Mater. Chem. A, 2015, 3, 19545 CAS
- H. Gu, C. Ma, J. Gu, J. Guo, X. Yan, J. Huang, Q. Zhang and Z. Guo, J. Mater. Chem. C, 2016, 4, 5890–5906 RSC
- V. Mittal, Macromol. Mater. Eng., 2014, 299, 906 CrossRef CAS
- E. S. Polsen, D. Q. McNerny, B. Viswanath, S. W. Pattinson and A. J. Hart, Sci. Rep., 2015, 5, 10257 CrossRef PubMed
- K. R. Paton, E. Varrla, C. Backes, R. J. Smith, U. Khan, A. O'Neill, C. Boland, M. Lotya, O. M. Istrate, P. King, T. Higgins, S. Barwich, P. May, P. Puczkarski, I. Ahmed, M. Moebius, H. Pettersson, E. Long, J. Coelho, S. E. O'Brien, E. K. McGuire, B. M. Sanchez, G. S. Duesberg, N. McEvoy, T. J. Pennycook, C. Downing, A. Crossley, V. Nicolosi and J. N. Coleman, Nat. Mater., 2014, 13, 624 CrossRef CAS PubMed
- D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitskii, Z. Sun, A. Slesarev, L. B. Alemany, W. Lu and J. M. Tour, ACS Nano, 2010, 4, 4806 CrossRef CAS PubMed
- S. Stankovich, R. D. Piner, S.-T. Nguyen and R. S. Ruoff, Carbon, 2006, 44, 3342 CrossRef CAS
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS
- H. Gu, S. B. Rapole, Y. Huang, D. Cao, Z. Luo, S. Wei and Z. Guo, J. Mater. Chem. A, 2013, 1, 2011 CAS
- H. Gu, J. Guo, H. Wei, X. Yan, D. Ding, X. Zhang, Q. He, S. Tadakamalla, X. Wang, T. C. Ho, S. Wei and Z. Guo, J. Mater. Chem. C, 2015, 3, 8152 RSC
- H. Gu, J. Guo, Q. He, S. Tadakamalla, X. Ziang, X. Yan, Y. Huang, H. A. Colorado, S. Wei and Z. Guo, Ind. Eng. Chem. Res., 2013, 52, 7718 CrossRef CAS
- H. Gu, J. Guo, H. Wei, S. Guo, J. Liu, Y. Huang, M. A. Khan, X. Wang, D. P. Young, S. Wei and Z. Guo, Adv. Mater., 2015, 27, 6277 CrossRef CAS PubMed
- H. Gu, S. Tadakamalla, Y. Huang, H. A. Colorado, Z. Luo, N. Haldolaarachchige, D. P. Young, S. Wei and Z. Guo, ACS Appl. Mater. Interfaces, 2012, 4, 5613 CAS
- Y.-J. Wan, L.-C. Tang, D. Yan, L. Zhao, Y. B. Li, L.-B. Wu, J.-X. Jiang and G.-Q. Lai, Compos. Sci. Technol., 2013, 82, 60 CrossRef CAS
- R. K. Agrawalla, S. Paul, P. K. Sahoo, A. K. Chakraborty and A. K. Mitra, J. Appl. Polym. Sci., 2015, 132, 41692 Search PubMed
- A. K. Chakraborty, V. R. Dhanak and K. S. Coleman, Nanotechnology, 2009, 20, 155704 CrossRef PubMed
- A. Suri, A. K. Chakraborty and K. S. Coleman, Chem. Mater., 2008, 20, 1705 CrossRef CAS
- K. S. Coleman, A. K. Chakraborty, S. R. Bailey, J. Sloan and M. Alexander, Chem. Mater., 2007, 19, 1076 CrossRef CAS
- V. Meriga, V. Sreeramulu, S. Sundaresan, C. Cahill, V. R. Dhanak and A. K. Chakraborty, J. Appl. Polym. Sci., 2015, 132, 42766 CrossRef
- M. A. Rafiee, J. Rafiee, J. Wang, H. Song, Z.-Z. Yu and N. Koratkar, ACS Nano, 2009, 3, 3884 CrossRef CAS PubMed
- F. Yavari, M. A. Rafiee, J. Rafiee, Z.-Z. Yu and N. Koratkar, ACS Appl. Mater. Interfaces, 2010, 2, 2738 CAS
- A. Zandiatashbar, C. R. Picu and N. Koratkar, Small, 2012, 8, 1676 CrossRef CAS PubMed
- M. A. Rafiee, J. Rafiee, I. Srivastava, Z. Wang, H. Song, Z.-Z. Yu and N. Koratkar, Small, 2010, 6, 179 CrossRef CAS PubMed
- M. M. Gudarzi and F. Sharif, eXPRESS Polym. Lett., 2012, 6, 1017 CrossRef
- Y.-J. Wan, L.-C. Tang, L.-X. Gong, D. Yan, Y.-B. Li, L.-B. Wu, J.-X. Jiang and G.-Q. Lai, Carbon, 2014, 69, 467 CrossRef CAS
- F. H. Gozny, M. H. G. Wichmann, B. Feidler and K. Schulte, Compos. Sci. Technol., 2005, 65, 2300 CrossRef
- V. Leon, R. Parret, R. Almairac, L. Alvarez, Z. Z. Zahab, J.-L. Bantignies, M.-R. Babaa, B. P. Doyle, P. Ienny and P. Parent, Carbon, 2012, 50, 4987 CrossRef CAS
- P.-C. Ma, Y.-S. Mo, B.-Z. Tang and J.-K. Kim, Carbon, 2010, 48, 1824 CrossRef CAS
- F. H. Gozny, M. H. G. Wichmann, U. Kopke, B. Fiedler and K. Schulte, Compos. Sci. Technol., 2004, 64, 2363 CrossRef
- W. Zhang, I. Srivastava, Y.-F. Zhu, C. R. Picu and N. Koratkar, Small, 2009, 5, 1403 CrossRef CAS PubMed
- F. H. Gozny, J. Nastalczyk, Z. Roslaniec and K. Schulte, Chem. Phys. Lett., 2003, 370, 820 CrossRef
- S. Niyogi, E. Bekyarova, M. E. Itkis, J. L. McWilliams, M. A. Hammon and R. C. Haddon, J. Am. Chem. Soc., 2006, 128, 7720 CrossRef CAS PubMed
- A. B. Bourlinos, D. Gournis, D. Petridis, T. Szabo, A. Szeri and I. Dekany, Langmuir, 2008, 19, 6050 CrossRef
- W. Li, X.-Z. Tang, H.-B. Zhang, Z.-G. Jiang, Z.-Z. Yu, X.-S. Du and Y.-W. Mai, Carbon, 2011, 49, 4724 CrossRef CAS
- A. K. Chakraborty, T. Plyhm, M. Barbezat, A. Necola and G. P. Terrasi, J. Nanopart. Res., 2011, 13, 6493 CrossRef CAS
- G. Gkikas and A. V. Paipetis, Meccanica, 2015, 50, 461 CrossRef
- H.-C. Hsu, I. Shown, H.-Y. Wei, Y.-C. Chang, H.-Y. Du, Y.-G. Lin, C.-A. Tseng, C.-H. Wang, L.-C. Chen, Y.-C. Lin and K.-H. Chen, Nanoscale, 2013, 5, 262 RSC
- O. C. Compton, D. A. Dikin, K. W. Putz, L. C. Brinson and S. T. Nguyen, Adv. Mater., 2010, 22, 892 CrossRef CAS PubMed
- G. S. Kumar, R. Roy, D. Sen, U. K. Ghorai, R. Thapa, N. Mazumder, S. Saha and K. K. Chattopadhyay, Nanoscale, 2014, 6, 3384 RSC
- L. G. Wade Jr, Organic Chemistry, Pearson Prentice Hall Inc., New Jersey, 2006 Search PubMed
- J. Lin, Y. Liu and C.-P. Wong, Langmuir, 2010, 26, 16110 CrossRef PubMed
- F. Tuinstra and J. L. Koenig, J. Chem. Phys., 1970, 53, 1126 CrossRef CAS
- A. C. Ferrari and J. Robertson, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 14095 CrossRef CAS
- J. I. Paredes, S. Villar-Rodil, A. Martínez-Alonso and J. M. D. Tascón, Langmuir, 2008, 24, 10560 CrossRef CAS PubMed
- A. M. Shamugharaj, J. H. Yoon, J. W. Yang and S. H. Ryu, J. Colloid Interface Sci., 2013, 40, 148 CrossRef PubMed
- C. May, Epoxy Resins: Chemistry and Technology, Marcel Dekker Inc., New York, 1988 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra12844k |
| This journal is © The Royal Society of Chemistry 2016 |