
A tumor-targeting drug delivery system based on cyclic NGR-modified, combretastatin A4-loaded, functionalized graphene oxide nanosheets
Abstract
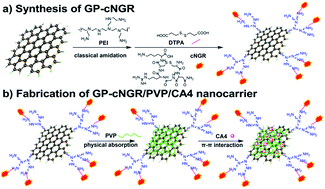
Graphene oxide has shown great potential in drug delivery. In this study, we developed a novel tumor-targeting drug carrier based on aminopeptidase N (APN or CD13)-targeting peptide (NGR) functionalized graphene oxide (GP–cNGR/PVP). For the preparation, graphene oxide (GO) was functionalized using polyethylenimine (PEI) covalently linked with cyclic NGR (cNGR), and then the nanosheets were conjugated with polyvinylpyrrolidone (PVP) via non-covalent interactions. The results showed that an efficient loading of combretastatin A4 (CA4) on GP–cNGR/PVP (0.5630 ± 0.0132 mg mg−1) was obtained. In vitro cytotoxicity and cellular uptake studies using two tumor cells (HT-1080, MCF-7) indicated that the GP–cNGR/PVP nanosheets could recognize certain tumor cells and enhance the uptake by cells, especially for cells overexpressing CD13 receptors. These results demonstrate that GP–cNGR/PVP could be a potential vehicle to delivery anticancer agents for specific cancer therapy.
 Please wait while we load your content...
Please wait while we load your content...

