
Collective formal synthesis of (±)-rhynchophylline and homologues†
Abstract
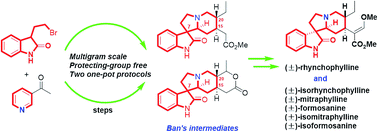
A collective formal synthesis approach to bioactive oxindole alkaloids, including (±)-rhynchophylline, (±)-isorhynchophylline, (±)-mitraphylline, (±)-formosanine, (±)-isomitraphylline, and (±)-isoformosanine, is completed in a protecting-group free manner. Besides multigram-scaled operations, the notable feature of the synthesis is the application of two one-pot, sequential transformations. Namely, two key tetracyclic intermediates pyridinium salt 9 and monoester 14 were prepared by a one-pot N-alkylation/cross-dehydrogenative coupling sequence and a one-pot Michael/Karpocho sequence with minimal purification, respectively.
- This article is part of the themed collection: Elegant Synthetic Routes to Indole Derivatives
 Please wait while we load your content...
Please wait while we load your content...

