
MOF-253-Pd(OAc)2: a recyclable MOF for transition-metal catalysis in water†
Ryan Van Zeeland‡
a,
Xinle Li‡ab,
Wenyu Huang*ab and
Levi M. Stanley*a
aDepartment of Chemistry, Iowa State University, Ames, IA 50011, USA. E-mail: whuang@iastate.edu; lstanley@iastate.edu
bAmes Laboratory, U.S. Department of Energy, Ames, IA 50011, USA
First published on 6th June 2016
Abstract
We report palladium(II)-functionalized MOF-253 (MOF-253-Pd(OAc)2) as a recyclable catalyst to form all-carbon quaternary centers via conjugate additions of arylboronic acids to β,β-disubstituted enones in aqueous media. We demonstrate MOF-253-Pd(OAc)2 can be reused 8 times to form ketone products in yields above 75% while maintaining its crystallinity. Additions of a range of stereoelectronically diverse arylboronic acids to a variety of β,β-disubstituted enones catalyzed by MOF-253-Pd(OAc)2 occur in modest-to-high yields (34–95%).
The ability to carry out chemical reactions in the most efficient, economical, and environmentally responsible manner is critical to a sustainable chemical enterprise.1 To this end, synthetic chemists have sought to develop processes that minimize hazardous reagents while increasing atom economy and energy efficiency.2 In many reactions, organic solvents constitute the majority of the chemical matter and are the primary source of hazardous waste.3 As a result, studies to replace hazardous organic solvents with more environmentally benign aqueous media have become a priority.4
Homogeneous transition-metal catalysts with outstanding activity and selectivity have been developed as a means to improve atom economy and energy efficiency in a broad range of reactions.5 Unlike classical heterogeneous catalysts, which often lack the activity and selectivity of their homogeneous counterparts, homogeneous transition-metal catalysts are often difficult or impossible to recover and reuse.6 Metal–organic frameworks (MOFs) have emerged as a promising platform for catalysis at the interface of traditional homogeneous and heterogeneous catalysis.7 In recent years, MOFs containing 2,2′-bipyridyl linker units (bpy-MOFs) have been shown to be capable of supporting a variety of transition-metal complexes. The utility of these transition metal-functionalized bpy-MOFs as recyclable catalysts has been demonstrated in a wide variety of organic transformations including: cross-coupling reactions;8 reductions of carbon dioxide9 and alkenes;10 oxidations of alcohols,11 water,9b,12 arylboronic acids,13 and alkenes;14 oxidative coupling of amines;9b C–H borylation and silylation of arenes;10,15 hydrosilylation of ketones;15a and hydroborations10 of alkenes, aldehydes and ketones.16 These transformations are typically carried out in aprotic reaction media and the structures of the bpy-MOFs are known to be relatively stable even at high temperatures. Much less is understood about the structural stability of bpy-MOFs and metalated derivatives in polar, protic solvents, especially at elevated reaction temperatures.17 As a result, metal-functionalized bpy-MOFs as catalysts of reactions run in polar, protic solvents remain underexplored.
We recently reported palladium(II) complexes of 2,2′-bipyridine that catalyse conjugate additions of arylboronic acids to β,β-disubstituted enones in aqueous media.18 However, the palladium species formed upon completion of the reaction cannot be reused as the catalyst. The ability of MOFs to support active metal complexes and prevent bimolecular deactivation processes9b,12,19 prompted us to study palladium(II)-functionalized bpy-MOFs as potentially recyclable catalysts for these conjugate addition reactions and a platform for green catalysis in water. Herein, we report studies to develop palladium(II)-functionalized bpy-MOFs as recyclable catalysts to form all-carbon quaternary centres via conjugate additions of arylboronic acids to β,β-disubstituted enones in aqueous media (Scheme 1).
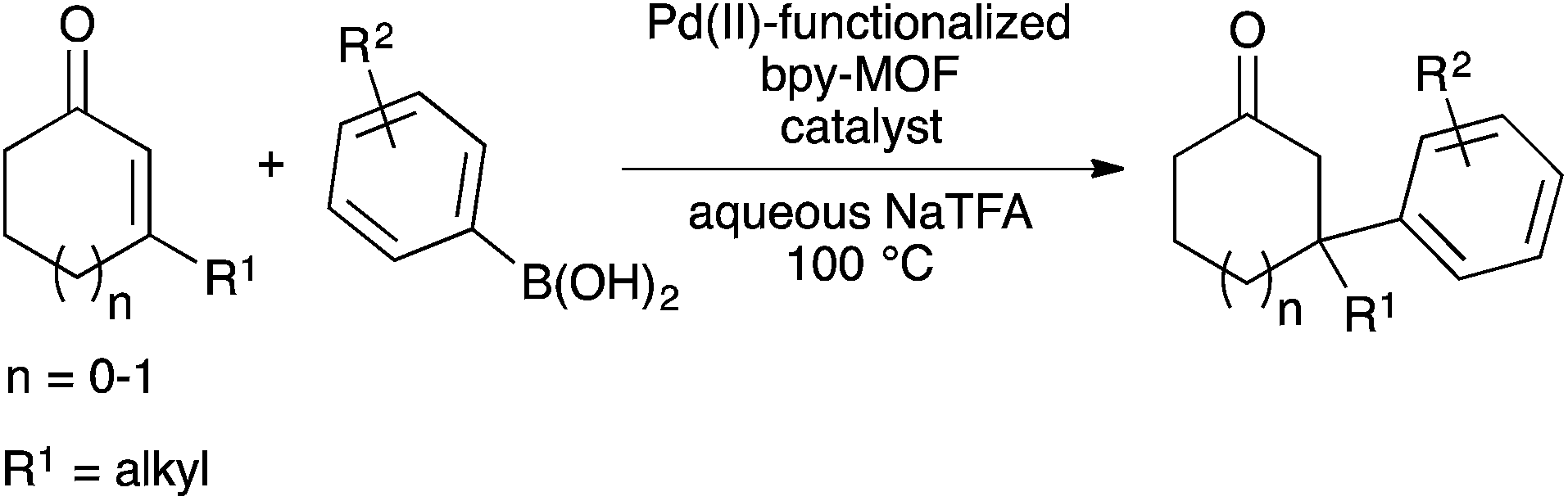 | ||
| Scheme 1 Formation of all-carbon quaternary centres via conjugate addition in aqueous media catalysed by Pd(II)-functionalized bpy-MOFs. | ||
MOF-253 and bpy-UiO-67, two prototypical MOFs containing 2,2′-bipyridyl linkers, were synthesized via reported protocols from [2,2′-bipyridine]-5,5′-dicarboxylic acid and either ZrCl4 or AlCl3·6H2O.20 Powder X-ray diffraction (PXRD) patterns and nitrogen physisorption analyses of the bpy-MOFs are consistent with reported data (Fig. S1–S4†). The postsynthetic metalation of bpy-UiO-67 and MOF-253 was performed by treating the bpy-MOFs with Pd(OAc)2 in acetone at ambient temperature to afford bpy-UiO-67-Pd(OAc)2 (C1) and MOF-253-Pd(OAc)2 (C2) (Fig. 1). The integrity of the bpy-MOFs was maintained after metalation based on PXRD patterns (Fig. S1 and S2†). The palladium content of the catalysts was determined quantitatively by inductively coupled plasma-mass spectrometry (ICP-MS).
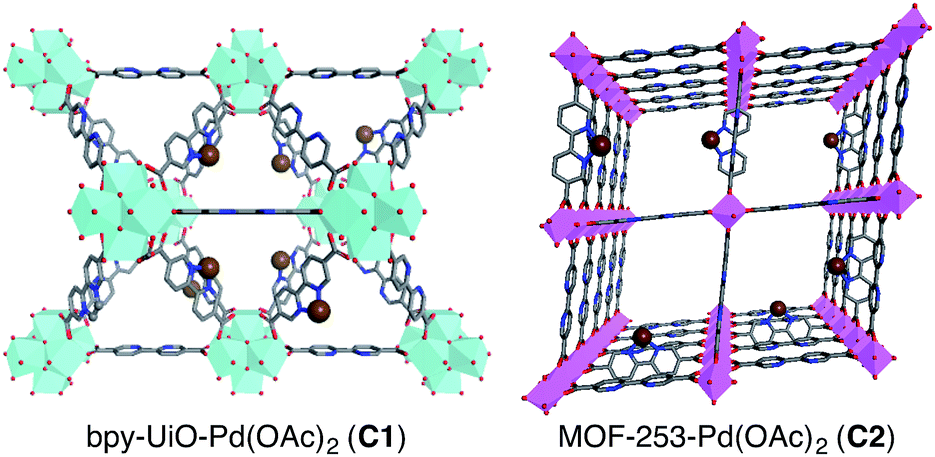 | ||
| Fig. 1 Idealized structures of bpy-UiO-67-Pd(OAc)2 (C1) and MOF-253-Pd(OAc)2 (C2). Cyan and pink octahedra represent Zr and Al clusters, respectively, while brown, red, blue, and gray spheres represent Pd(OAc)2 species, O, N, and C atoms, respectively; H atoms are omitted for clarity. | ||
With bpy-UiO-67-Pd(OAc)2 and MOF-253-Pd(OAc)2 in hand, we studied the model reaction of phenylboronic acid with 3-methylcyclohex-2-en-1-one 1a to evaluate the utility of these Pd(II)-functionalized MOFs as catalysts in polar, protic media (Table 1). The reaction of enone 1a with 1.2 equivalents of phenylboronic acid in the presence of bpy-UiO-67-Pd(OAc)2 (C1) (1.5 mol% total palladium based on 1a) did not form ketone 2a at 60 °C and formed 2a in 2% yield at 80 °C when the reactions were run in methanol (entries 1 and 2). Changing the reaction medium from methanol to 50 mM aqueous sodium trifluoroacetate (aq. NaTFA, pH = 8.2) led to the formation of ketone 2a in 20% yield when the reaction was run at 80 °C (entry 3). Ketone 2a was formed in 50% yield upon increasing the reaction temperature to 100 °C (entry 4).
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst (mol% Pd) | wt% Pd in MOFb | Temp (°C) | PhB(OH)2 (equiv.) | Yieldc (%) |
| a Reaction conditions: 1a (0.500 mmol), PhB(OH)2 (0.600–1.00 mmol), MOF catalyst (0.008–0.013 mmol Pd), reaction medium (0.33 mL), 16 h.b Weight% Pd loaded in the MOF determined by ICP-MS.c Determined by 1H NMR spectroscopy using dibromomethane as an internal standard.d Reaction run in methanol.e Reaction run for 2 h. | |||||
| 1d | C1 (1.5) | 5.0 | 60 | 1.2 | 0 |
| 2d | C1 (1.5) | 5.0 | 80 | 1.2 | 2 |
| 3 | C1 (1.5) | 5.0 | 80 | 1.2 | 20 |
| 4 | C1 (1.5) | 5.0 | 100 | 1.2 | 50 |
| 5 | C1 (2.5) | 5.0 | 100 | 1.2 | 74 |
| 6 | C1 (2.5) | 5.0 | 100 | 2.0 | 90 |
| 7e | C1 (2.5) | 8.1 | 100 | 2.0 | 99 |
| 8e | C2 (2.5) | 8.4 | 100 | 2.0 | 99 |
| 9e | Bpy-UiO-67 (0.0) | 0.0 | 100 | 2.0 | 0 |
| 10e | UiO-67-Pd(OAc)2 (2.4) | 2.4 | 100 | 2.0 | 0 |
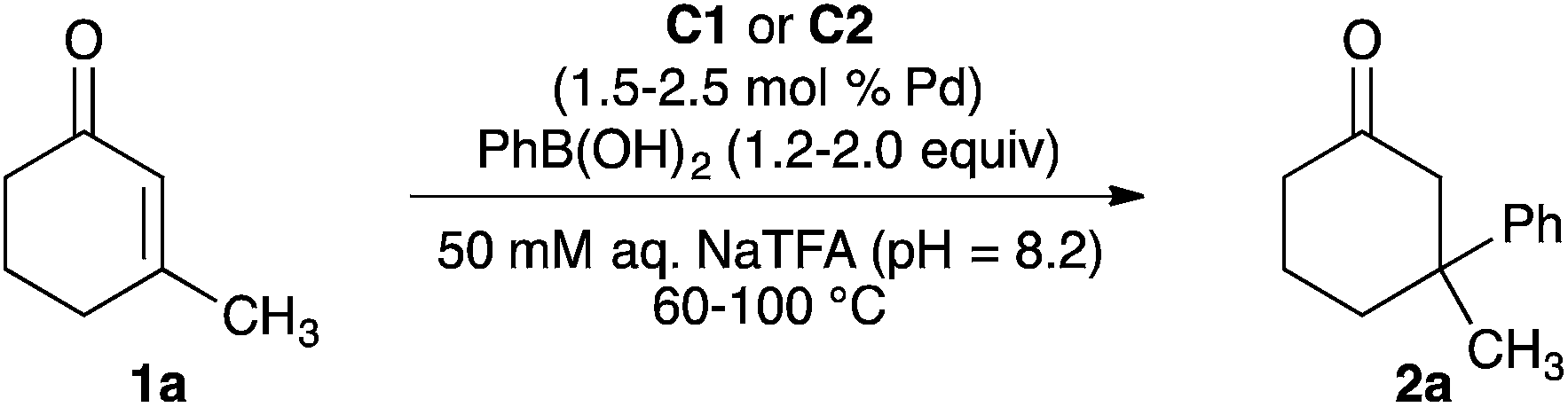
We found that the total loading of palladium in the reaction, the number of equivalents of phenylboronic acid, and the weight percentage of palladium present in the MOF significantly impact the yield of our model reaction (entries 5–7). Increasing the total palladium content of the reaction from 1.5 mol% to 2.5 mol% and the amount of phenylboronic acid from 1.2 equivalents to 2.0 equivalents led to the formation of 2a in 90% yield (entry 6). When the weight% Pd loaded in the MOF was increased from 5.0% to 8.1%, the reaction was complete after two hours and generated 2a in 99% yield (entry 7). The reaction of phenylboronic acid with 1a catalysed by MOF-253-Pd(OAc)2 C2 in place of C1 also formed 2a in 99% yield in two hours (entry 8). Reactions of 1a with phenylboronic acid run in the presence of bpy-UiO-67 with no palladium and an analogous MOF without bipyridine sites, UiO-67-Pd(OAc)2, did not occur to form the conjugate addition product 2a (entries 9 and 10). These results are consistent with supported Pd(II)–bipyridine complexes as the active catalysts when MOFs C1 and C2 are used to promote the model reaction.
The palladium-functionalized MOFs bpy-UiO-67-Pd(OAc)2 C1 and MOF-253-Pd(OAc)2 C2 perform similarly as catalysts in the model reaction. Two features of MOF-253-Pd(OAc)2 C2 led us to select this material for additional catalytic studies. The larger pore size of MOF-253![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 21 compared to bpy-UiO-6722 is attractive because the resulting larger pore volumes will be able to accommodate a wider array of enone and arylboronic acid substrates and will facilitate flux of reagents and products into and out of the pores. In addition, we found that MOF-253 could be consistently metalated with approximately 8 weight% Pd, while we observed significant batch-to-batch variations for metalation of bpy-UiO-67 with Pd(OAc)2.
21 compared to bpy-UiO-6722 is attractive because the resulting larger pore volumes will be able to accommodate a wider array of enone and arylboronic acid substrates and will facilitate flux of reagents and products into and out of the pores. In addition, we found that MOF-253 could be consistently metalated with approximately 8 weight% Pd, while we observed significant batch-to-batch variations for metalation of bpy-UiO-67 with Pd(OAc)2.
To gain insight into the stability of MOF-253-Pd(OAc)2 C2 under our aqueous reaction conditions, we evaluated the recyclability of C2 in our model addition of phenylboronic acid to enone 1a (Scheme 2). Consistent with our data in Table 1, the initial reaction with pristine C2 as catalyst formed ketone 2a in 99% yield in less than two hours. Upon recovery of the catalyst and exposure to additional enone 1a, phenylboronic acid, and aqueous NaTFA, the yield of ketone 2a dropped to 92% in run 2 and 76–80% in runs 3 and 4 after 2–2.5 hour reaction times. The yield of 2a could be increased to 90–99% for runs 5–9 by extending the reaction times.
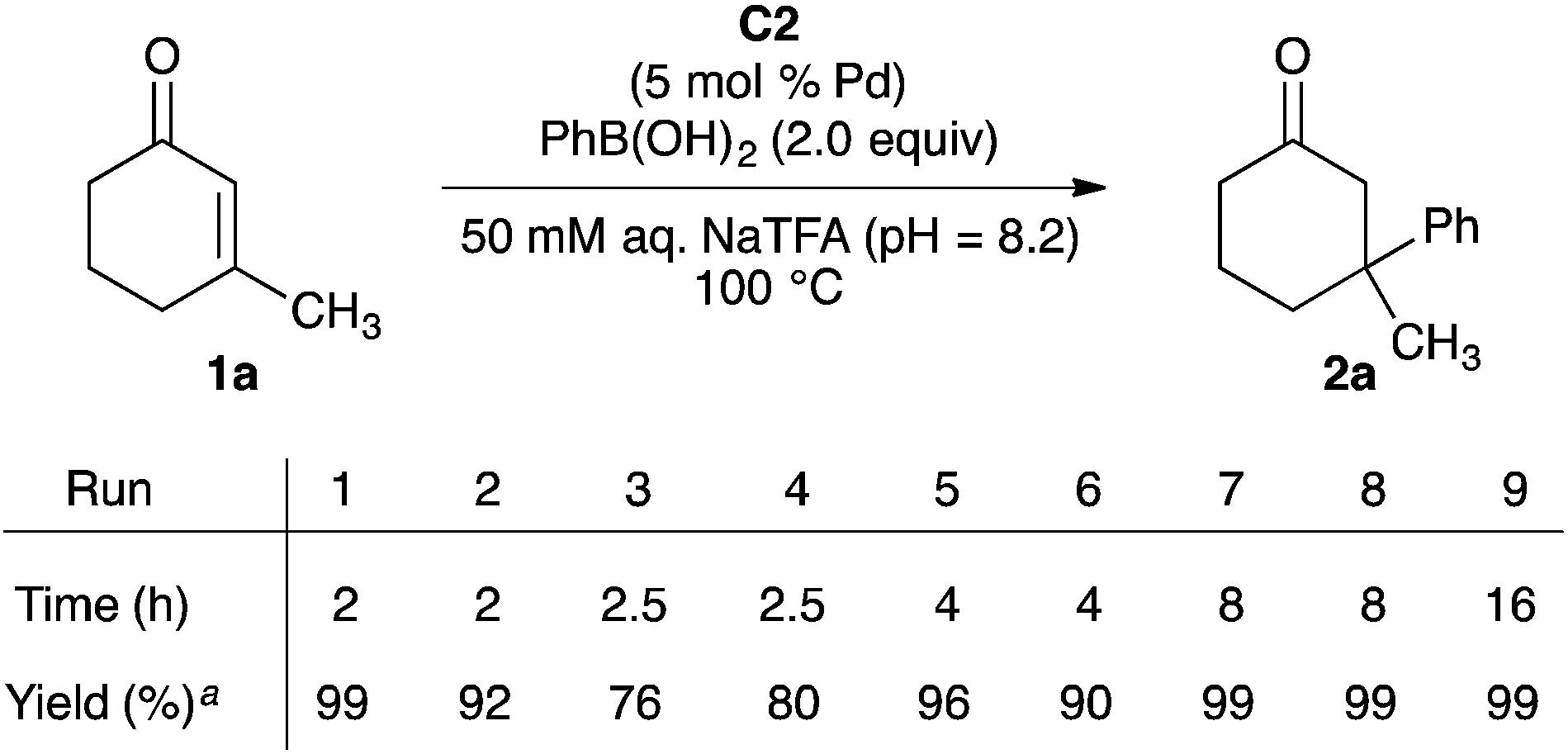 | ||
| Scheme 2 Recycling of C2 in the addition of PhB(OH)2 to enone 1a. Reaction conditions: 1a (1.50 mmol, 1.00 equiv.), PhB(OH)2 (3.00 mmol, 2.00 equiv.), C2 (0.075 mmol, 0.050 equiv.) and aqueous 50 mM NaTFA (1.00 mL). aDetermined by 1H NMR spectroscopy using dibromomethane as an internal standard. | ||
The results shown in Scheme 2 clearly demonstrate the ability to reuse MOF-253-Pd(OAc)2 C2 in conjugate additions of phenylboronic acid to enone 1a. However, these results also show that, at minimum, partial degradation of the C2 occurs over time. To verify that the solid MOF-supported 2,2′-bipyridine complex of Pd(OAc)2 is the active catalyst in these reactions, we performed leaching tests to eliminate the possibility for MOF degradation into an active and homogeneous palladium species (see ESI†). Exposure of C2 to the reagents and reaction conditions for two hours and analysis of the palladium content of the aqueous supernatant showed that 0.6% of the palladium had leached out of C2. However, the palladium found in the supernatant is not catalytically competent under our reaction conditions. Ketone 2a is formed in 1% yield after two hours when the palladium contained in the supernatant is evaluated as a catalyst of the model reaction under the optimized reaction conditions.
To develop an understanding of the rate of catalyst deactivation under our reaction conditions, we conducted an additional recycling study where C2 was reused and the yield of the conjugate addition reaction was determined after one-hour reaction times (Fig. 2). As expected from our initial recycling experiment, a slow decline in the activity of the catalyst is observed during the initial recycling runs. By the fourth run, the activity of the catalyst is halved and ketone 2a is formed in 29% yield compared to 60% in the first run. However, the activity of the catalyst remains consistent in runs 4–10 and 2a is formed in 23–30% yield after one hour.
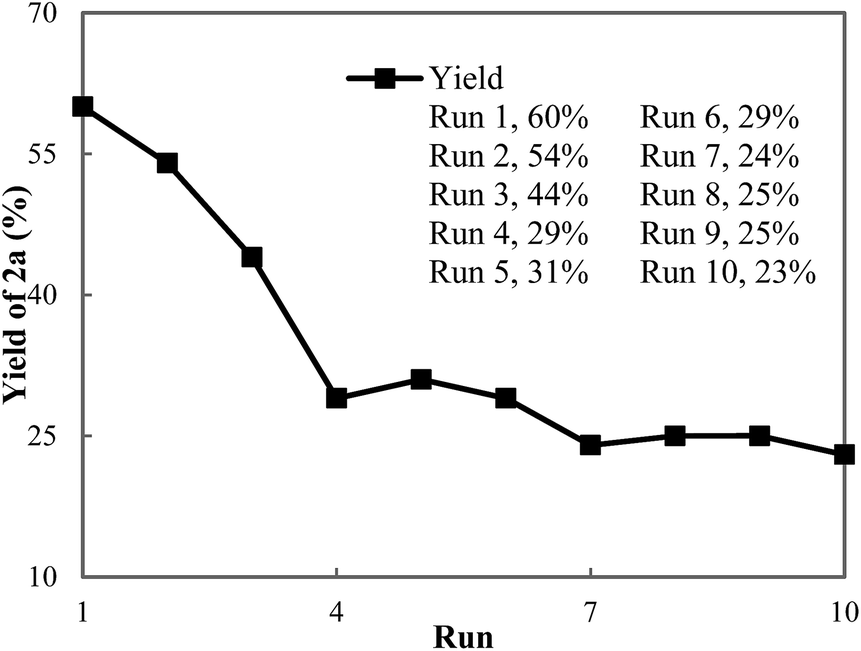 | ||
| Fig. 2 Low conversion recycling experiments. Reaction conditions: 1a (1.50 mmol, 1.00 equiv.), PhB(OH)2 (3.00 mmol, 2.00 equiv.), C2 (0.075 mmol, 0.050 equiv.), and aqueous 50 mM NaTFA (1.0 mL, pH = 8.2), 1 h reaction time. Yields of 2a were determined by 1H NMR spectroscopy using dibromomethane as an internal standard. | ||
The PXRD pattern of MOF-253-Pd(OAc)2 C2 after the 10th run remained unchanged from that of C2 before catalysis (Fig. S5†). More importantly, the used C2 possesses comparably high surface area relative to freshly prepared C2 (Fig. S6†). However, the decrease in activity of the catalyst after two hours (run 2 in Fig. 2) is not consistent with the quantity of palladium lost to leaching over two hours. The combination of these results suggests additional pathways for deactivation of the palladium catalyst are operative.
With a robust palladium-functionalized MOF for catalysis in water identified, we evaluated the scope of the conjugate addition reaction. Additions of a variety of arylboronic acids to a selection of enones 1a–c catalysed by MOF-253-Pd(OAc)2 C2 are summarized in Scheme 3. As demonstrated in our optimization studies, the addition of phenylboronic acid to 3-methylcyclohex-2-en-1-one 1a occurs to form ketone 2a in 90% yield. Additions of 4-substituted arylboronic acids containing electron-donating, electron-withdrawing, and halogen substituents to enone 1a formed ketones 2b–d in 80–90%. 3-Substituted arylboronic acids are also suitable substrates for conjugate additions catalysed by C2. Additions of 3-methoxy- and 3-chlorophenylboronic acids to enone 1a generated ketones 2e and 2f in 91% yield. The addition of 2-methoxyphenylboronic acid to 1a occurred to form ketone 2g in 34% yield. However, additions of 2-substituted arylboronic acid lacking a strong electron-donating substituent did not form conjugate addition products. In these cases, protodeborylation of the 2-substituted arylboronic acid is the primary reaction pathway observed.23
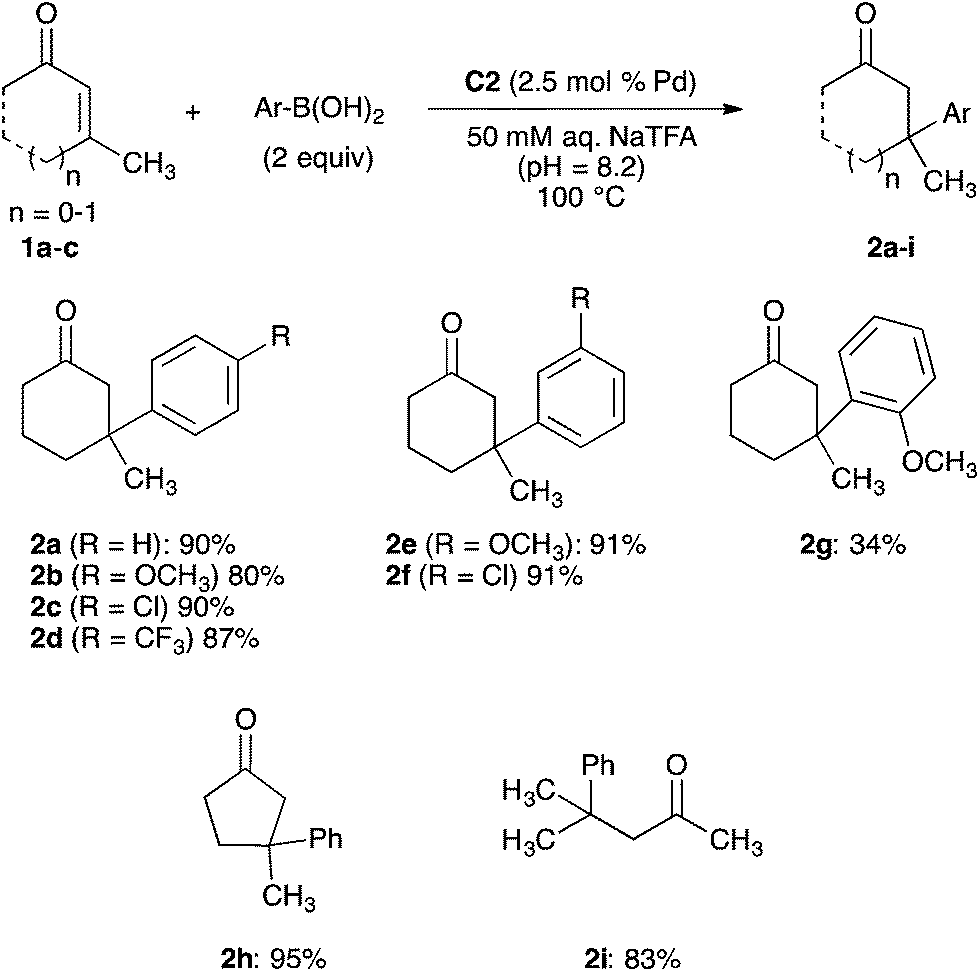 | ||
| Scheme 3 Conjugate addition of arylboronic acids to enones 1a–c catalysed by MOF-253-Pd(OAc)2 C2. Reaction conditions: 1a–c (0.500 mmol), arylboronic acid (1.00 mmol), MOF-253-Pd(OAc)2 C2 (0.013 mmol of Pd), 50 mM aqueous NaTFA (0.33 mL, pH = 8.2), 100 °C, 2–18 h. Isolated yields are reported after purification by flash column chromatography. | ||
MOF-253-Pd(OAc)2 C2 also catalyses conjugate additions of phenylboronic acid to additional cyclic and acyclic β,β-disubstituted enones. The addition of phenylboronic acid to 3-methylcyclopent-2-en-1-one 1b forms ketone 2h in 95% yield. The addition of phenylboronic acid to acyclic 4-methylpent-3-en-2-one 1c generated ketone 2i in 83% yield. However, additions of phenylboronic acid to 3-arylcyclohex-2-en-1-ones and 3-methylcyclohept-2-en-1-one occurred in <10% yield in the presence of MOF-253-Pd(OAc)2 C2. The poor reactivity of these two enone substrates is consistent with analogous reactions carried out in aqueous media and catalysed by a complex of 2,2′-bipyridine and palladium trifluoroacetate that require higher catalyst and arylboronic acid loadings. Attenuated rates for catalysis by C2 in combination with rates of protodeborylation that remain consistent regardless of the identity of the catalyst can lead to arene formation through protodeborylation as the primary reaction pathway for these more challenging substrates classes.
Conclusions
In summary, we have established two palladium(II)-functionalized bpy-MOFs, bpy-UiO-67-Pd(OAc)2 and MOF-253-Pd(OAc)2, as competent catalysts for conjugate additions of arylboronic acids to β,β-disubstituted enones in water. We have also demonstrated that MOF-253-Pd(OAc)2 is a reusable catalyst system that promotes additions of a range of arylboronic acid to β,β-disubstituted enone reaction partners to form ketones containing quaternary carbon centres. The development of MOF-253-Pd(OAc)2 as a platform for transition-metal catalysis in water sets the stage for new applications of this and related catalyst systems in the areas of green catalysis. Studies to improve the catalytic activities and stabilities of MOF-253-Pd(OAc)2 and additional metalated derivatives for new catalytic transformations in aqueous environments are on-going.Acknowledgements
We thank Iowa State University, the Iowa State University Center for Catalysis, and the Ames Laboratory for supporting this work. The Ames Laboratory is operated for the U.S. Department of Energy by Iowa State University under contract number DE-AC02-07CH11358. We thank Professor Gordon J. Miller (Iowa State University) for use of PXRD instrumentation in his group.Notes and references
- (a) C. H. Christensen and J. K. Norskov, Science, 2010, 327, 278–279 CrossRef PubMed; (b) M. Poliakoff and P. Licence, Nature, 2007, 450, 810–812 CrossRef CAS PubMed; (c) G. A. Somorjai, A. M. Contreras, M. Montano and R. M. Rioux, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10577–10583 CrossRef CAS PubMed.
- M. B. Gawande, V. D. B. Bonifacio, R. Luque, P. S. Branco and R. S. Varma, Chem. Soc. Rev., 2013, 42, 5522–5551 RSC.
- D. J. C. Constable, C. Jimenez-Gonzalez and R. K. Henderson, Org. Process Res. Dev., 2007, 11, 133–137 CrossRef CAS.
- (a) J. M. Crow, Chem. World, 2008, 5, 62–65 Search PubMed; (b) C. J. Li, Chem. Rev., 2005, 105, 3095–3165 CrossRef CAS PubMed.
- (a) G. E. Dobereiner and R. H. Crabtree, Chem. Rev., 2010, 110, 681–703 CrossRef CAS PubMed; (b) G. C. Fortman and S. P. Nolan, Chem. Soc. Rev., 2011, 40, 5151–5169 RSC; (c) D. J. Gorin, B. D. Sherry and F. D. Toste, Chem. Rev., 2008, 108, 3351–3378 CrossRef CAS PubMed; (d) S. Z. Tasker, E. A. Standley and T. F. Jamison, Nature, 2014, 509, 299–309 CrossRef CAS PubMed.
- D. J. Cole-Hamilton, Science, 2003, 299, 1702–1706 CrossRef CAS PubMed.
- (a) A. H. Chughtai, N. Ahmad, H. A. Younus, A. Laypkov and F. Verpoort, Chem. Soc. Rev., 2015, 44, 6804–6849 RSC; (b) A. Dhakshinamoorthy and H. Garcia, Chem. Soc. Rev., 2012, 41, 5262–5284 RSC; (c) K. Leus, Y. Y. Liu and P. Van Der Voort, Catal. Rev., 2014, 56, 1–56 CrossRef CAS; (d) M. Yoon, R. Srirambalaji and K. Kim, Chem. Rev., 2012, 112, 1196–1231 CrossRef CAS PubMed; (e) S. T. Madrahimov, J. R. Gallagher, G. Zhang, Z. Meinhart, S. J. Garibay, M. Delferro, J. T. Miller, O. K. Farha, J. T. Hupp and S. T. Nguyen, ACS Catal., 2015, 5, 6713–6718 CrossRef CAS; (f) C. M. McGuirk, M. J. Katz, C. L. Stern, A. A. Sarjeant, J. T. Hupp, O. K. Farha and C. A. Mirkin, J. Am. Chem. Soc., 2015, 137, 919–925 CrossRef CAS PubMed.
- (a) C. H. Bai, S. P. Jian, X. F. Yao and Y. W. Li, Catal. Sci. Technol., 2014, 4, 3261–3267 RSC; (b) L. Y. Chen, Z. Q. Gao and Y. W. Li, Catal. Today, 2015, 245, 122–128 CrossRef CAS; (c) L. Y. Chen, S. Rangan, J. Li, H. F. Jiang and Y. W. Li, Green Chem., 2014, 16, 3978–3985 RSC; (d) H. H. Fei and S. M. Cohen, Chem. Commun., 2014, 50, 4810–4812 RSC; (e) M. Wang, B. Z. Yuan, T. M. Ma, H. F. Jiang and Y. W. Li, RSC Adv., 2012, 2, 5528–5530 RSC.
- (a) D. R. Sun, Y. H. Gao, J. L. Fu, X. C. Zeng, Z. N. Chen and Z. H. Li, Chem. Commun., 2015, 51, 2645–2648 RSC; (b) C. Wang, Z. G. Xie, K. E. deKrafft and W. L. Lin, J. Am. Chem. Soc., 2011, 133, 13445–13454 CrossRef CAS PubMed.
- T. Zhang, K. Manna and W. B. Lin, J. Am. Chem. Soc., 2016, 138, 3241–3249 CrossRef CAS PubMed.
- F. Carson, S. Agrawal, M. Gustafsson, A. Bartoszewicz, F. Moraga, X. D. Zou and B. Martin-Matute, Chem.–Eur. J., 2012, 18, 15337–15344 CrossRef CAS PubMed.
- C. Wang, J. L. Wang and W. B. Lin, J. Am. Chem. Soc., 2012, 134, 19895–19908 CrossRef CAS PubMed.
- X. Yu and S. M. Cohen, Chem. Commun., 2015, 51, 9880–9883 RSC.
- T. Toyao, K. Miyahara, M. Fujiwaki, T. H. Kim, S. Dohshi, Y. Horiuchi and M. Matsuoka, J. Phys. Chem. C, 2015, 119, 8131–8137 CAS.
- (a) K. Manna, T. Zhang, F. X. Greene and W. B. Lin, J. Am. Chem. Soc., 2015, 137, 2665–2673 CrossRef CAS PubMed; (b) K. Manna, T. Zhang and W. B. Lin, J. Am. Chem. Soc., 2014, 136, 6566–6569 CrossRef CAS PubMed.
- For recent examples of palladium-functionalized MOFs containing linker units other than 2,2′-bipyridines, see: (a) J. W. Brown, N. N. Jarenwattananon, T. Otto, J. L. Wang, S. Gloggler and L. S. Bouchard, Catal. Commun., 2015, 65, 105–107 CrossRef CAS; (b) S. A. Burgess, A. Kassie, S. A. Baranowski, K. J. Fritzsching, K. Schmidt-Rohr, C. M. Brown and C. R. Wade, J. Am. Chem. Soc., 2016, 138, 1780–1783 CrossRef CAS PubMed; (c) A. Dhakshinamoorthy, A. M. Asiri and H. Garcia, Chem. Soc. Rev., 2015, 44, 1922–1947 RSC; (d) B. Gui, K. K. Yee, Y. L. Wong, S. M. Yiu, M. Zeller, C. Wang and Z. T. Xu, Chem. Commun., 2015, 51, 6917–6920 RSC; (e) D. Saha, R. Sen, T. Maity and S. Koner, Langmuir, 2013, 29, 3140–3151 CrossRef CAS PubMed.
- J. B. DeCoste, G. W. Peterson, H. Jasuja, T. G. Glover, Y. G. Huang and K. S. Walton, J. Mater. Chem. A, 2013, 1, 5642–5650 CAS.
- R. Van Zeeland and L. M. Stanley, ACS Catal., 2015, 5, 5203–5206 CrossRef CAS.
- K. K. Tanabe, N. A. Siladke, E. M. Broderick, T. Kobayashi, J. F. Goldston, M. H. Weston, O. K. Farha, J. T. Hupp, M. Pruski, E. A. Mader, M. J. A. Johnson and S. T. Nguyen, Chem. Sci., 2013, 4, 2483–2489 RSC.
- (a) G. Nickerl, M. Leistner, S. Helten, V. Bon, I. Senkovska and S. Kaskel, Inorg. Chem. Front., 2014, 1, 325–330 RSC; (b) X. Qiu, C. Len, R. Luque and Y. W. Li, ChemSusChem, 2014, 7, 1684–1688 CrossRef CAS PubMed.
- P. Valvekens, E. D. Bloch, J. R. Long, R. Ameloot and D. E. De Vos, Catal. Today, 2015, 246, 55–59 CrossRef CAS.
- L. J. Li, S. F. Tang, C. Wang, X. X. Lv, M. Jiang, H. Z. Wu and X. B. Zhao, Chem. Commun., 2014, 50, 2304–2307 RSC.
- (a) C. L. Boeser, J. C. Holder, B. L. H. Taylor, K. N. Houk, B. M. Stoltz and R. N. Zare, Chem. Sci., 2015, 6, 1917–1922 RSC; (b) J. Buter, R. Moezelaar and A. Minnaard, Org. Biomol. Chem., 2014, 12, 5883–5890 RSC; (c) A. J. J. Lennox and G. C. Lloyd-Jones, Isr. J. Chem., 2010, 50, 664–674 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details and procedures, materials, and characterization data (nuclear magnetic resonance (NMR), powder X-ray diffraction patterns (PXRD), and nitrogen sorption measurements). See DOI: 10.1039/c6ra12746k |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2016 |