
d-α-Tocopheryl polyethylene glycol 1000 succinate conjugated folic acid nanomicelles: towards enhanced bioavailability, stability, safety, prolonged drug release and synergized anticancer effect of plumbagin
Abstract
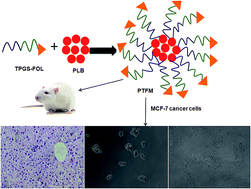
The aim of the study was to develop plumbagin (PLB) loaded D-α-tocopheryl polyethylene glycol 1000 succinate (vitamin E TPGS1k or TPGS)-folic acid conjugated nanomicelles (denoted as PTFM) to achieve controlled and targeted delivery with synergized anticancer potency and reduced PLB toxicity. PLB loaded TPGS micelles without folic acid conjugation (denoted as PTM) and PTFM were smaller in size and good encapsulation efficiency. The critical micellar concentration of TPGS-folic acid micellar solution was 0.015 mg ml−1. The micelles demonstrated sustained release and biocompatibility in hemolytic toxicity assay. Bioavailability of PLB from PTM and PTFM was increased by 3.9 and 4.8 fold with long circulation time, slower plasma elimination and no sign of blood and tissue toxicity as compared to free PLB. Moreover, formulated micelles demonstrated higher in vitro anticancer activity in folate over expressed human breast cancer MCF-7 cells. The targeting effect for the PTFM was also demonstrated. The concentration of the drug needed for growth inhibition of 50% of cells in a designed time period (GI50) was 13.15 ± 1.31 μg ml−1 for PLB while it was decreased by 40.68% for the PTM. Furthermore, the GI50 value of PTFM was 3.2 ± 0.4 μg ml−1, i.e., a 75.67% decrease was observed as compared to free PLB. A synergistic effect of TPGS and PLB was also achieved which reveals a new concept of a polymeric micellar drug delivery system where a carrier having therapeutic effects brings about a reduction in dose as well as cost.
 Please wait while we load your content...
Please wait while we load your content...

