
Facile synthesis of porous NiCo2O4 nanoflakes as magnetic recoverable catalysts towards the efficient degradation of RhB†
Abstract
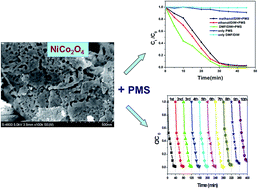
In this work, a class of flake-shaped magnetic NiCo2O4 material is fabricated by a facile hydrothermal reaction followed by a calcination treatment process. The prepared flake-like NiCo2O4 displays porous features that endow it with a large specific surface area (142.48 m2 g−1) and a narrow pore size distribution (3.70 nm). Furthermore, the NiCo2O4 samples were used as high-performance heterogeneous catalysts in the activation of peroxymonosulphate (PMS) to produce active radicals SO4−˙ and HO˙. Then, the produced SO4−˙ and HO˙ can further attack and degrade organic dyes. The catalytic results show that the magnetic flake-like NiCo2O4 catalyst can completely degrade rhodamine B (RhB) dye within 30 min with the assistance of PMS. In addition, the catalysts could be magnetically recovered, displayed high catalytic activity and excellent cycling stability. It is believed that the specific porous features, including high specific surface area and tailored pore size distributions, and surface defects can ensure the high activation of PMS for the catalytic oxidation of RhB. More importantly, the present synthetic method is facile, controllable and scalable, which highlights its potential in energy-storage, environmental treatment, and biology-related fields.
 Please wait while we load your content...
Please wait while we load your content...

