
The effect of charge alteration and flexibility on the function and structural stability of sweet-tasting brazzein
S. Shahrbanoo Jafaria,
Vahab Jafarian*a,
Khosrow Khalifehb,
Parisa Ghanavatiana and
S. Akram Shirdela
aLaboratory of Biochemistry, Department of Biology, Faculty of Sciences, University of Zanjan, Zanjan, Iran. E-mail: v.jafarian@znu.ac.ir
bLaboratory of Biophysics, Department of Biology, Faculty of Sciences, University of Zanjan, Zanjan, Iran
First published on 13th June 2016
Abstract
To identify the structural and functional roles of Glu9 located at the first loop of sweet-tasting brazzein, two different mutants (E9K and E9G) of the minor form of brazzein were designed and constructed. Upon histidine-tag (His-tag) elimination and purification of the protein variants, their activity and structural features were evaluated. According to the resulting data of taste evaluation, it was found that the sweetness of both mutants is raised, when compared with that of the wild-type (WT) protein. Intrinsic and ANS-based fluorescence spectra revealed that the structural compactness of the protein increases in both mutants. However, far-UV CD data indicated that the secondary structural content of the protein is not changed significantly upon mutation. Furthermore, the isothermal denaturation experiments using urea as a chemical denaturant showed that the E9K mutant has more conformational stability relative to the WT protein, while the structural stability of the E9G mutant is less than that of the WT protein. We concluded that changing the net charge of brazzein toward a positive one can influence the interaction of the protein with its corresponding receptors and leads to increasing its sweetness power.
Introduction
In the recent century, a number of sweet-tasting proteins were isolated from African and western Asian fruits.1 These carbohydrate-free compounds do not share structural properties with sucrose and other carbohydrates.2 However, they are approximately 1000 times sweeter than sucrose.3 So they can be used as suitable alternatives for artificial sweeteners such as sucralose, acesulfame K, saccharin and sodium cyclamate.4 It is worth mentioning that there are 8 known sweet-tasting proteins; among them, brazzein is the smallest one containing 53 and 54 residues in its minor and major forms, respectively.5–8 The combination of physicochemical properties of brazzein such as thermostability, high solubility and its tolerance to a wide range of pH makes it a good choice for both basic and applied research.9,10 The three-dimensional structure of brazzein by nuclear magnetic resonance spectroscopy (NMR) revealed that it contains a short α-helix comprising residues 21–29 and three anti-parallel β-strands including 1, 2 and 3 strands comprising residues 5–7, 33–39 and 44–50, respectively. These strands are spatially connected to each other by four disulfide bonds.11 Brazzein also has three flexible loops. The first one (residues 9–19) is a flexible loop between the first β-strand and α-helix. Moreover, it is one of the three popular interaction sites with the receptor.12 The second loop (residues 29–33) with saturated positive charges plays a central role in the sweetness of brazzein. The third loop including residues 39–44 was proposed as one of the three potential binding sites with T1R2 and T1R3 receptors.13,14Several researchers have tried to increase the sweetness power of brazzein. In line with these investigations some parts of brazzein have been reported as critical regions responsible for its sweetness.6 Assadi-Porter et al. identified a region between the N and C-terminal along with a flexible loop around Arg43 which serve as two important regions in the sweetness of brazzein.15 Additionally, Jin et al. reported that the positive charge of the residue at position 36 between two strands ranging from 29–33 and 39–43 alongside the C-terminal of the protein play essential roles for its sweetness.5 Moreover, according to other studies on several sweetener proteins such as thaumatin, monellin, hen egg white lysozyme, kurkulina and their representative mutants, a direct correlation was observed between sweetness and the number of their positive charges.5,16
In the current research, upon bioinformatics studies on the structure of brazzein, Glu9 located at the first loop of the protein was selected as the site of mutation and it was substituted with lysine (E9K) and glycine (E9G). The sweetness power of the protein variants was evaluated by means of a human taste panel. Circular dichroism (CD) in the far-UV region, intrinsic and ANS-based fluorescence as well as equilibrium denaturation experiments in various concentrations of urea were used for structural characterization of the wild-type and mutants.
Materials and methods
Materials
The following compounds and reagents were used for the current research: the markers for estimating the molecular weights of DNA (CinnaGen PR901645, Vivantis VC100 bp), IPTG (Fermentas, Germany), kanamycin, nucleotides and agarose (Invitrogen, Carlsbad, CA, USA), nickel-agarose (Qiagen, Hilden, Germany), Amicon-ultra-0.5 mL 3 kDa (Millipore, USA), Pfu DNA Polymerase (Thermo Scientific, USA) and plasmid extraction and purification kite for PCR products (Bioneer, Korea). Other chemicals were obtained from Merck Company (Germany).Bioinformatics
The three-dimensional structure of the mutants was made by the MODELLER program version 9v7,17 using the PDB structure of the WT protein (PDB ID: 4HE7) as a template. Among ten generated models, the final model was selected by evaluation tests of the DOPE (discrete optimized protein energy) score (https://modbase.compbio.ucsf.edu), Errat, Verify3D, and ProCheck (http://nihserver.mbi.ucla.edu/SAVS/) programs. The structure of the WT and mutants was further examined by a protein interaction calculator (PIC) server18 and Swiss-PDB Viewer program.19 The VADAR (volume, area, dihedral angle reporter) server was also used for investigating the accessibility of residues on the structure of the WT and mutants.20Description of molecular biology
The synthetic gene containing methionine in the N-terminal was inserted in the PET28a vector under the control of a T7 promoter (further information is not provided here). Upon designing the oligonucleotide primers, PCR was carried out using the Pfu enzyme. Before transformation, the elimination of template plasmids was performed by DpnI 10U in 37 °C for 16 hours. After clean-up of the PCR products, the products were transferred to competent cells (E. coli XL1-Blue) and the cells were chemically transformed.After sequencing of PET28a using the T7 promoter and T7 terminator and the evaluation of mutations with GeneRunner ver. 3.05. software, the sequence alignment of the WT protein with mutants was carried out by the CLUSTAL W program.
Expression and purification of the recombinant protein
For gene expression, recombinant genes of brazzein (both WT and mutants) were transferred to an E. coli strain SHuffle® T7 Express as Expression system. To prepare the pre-culture, a single colony of the bacteria was cultured in 10 mL LB medium containing kanamycin (50 μg mL−1) and then it was incubated at 30 °C. 2 mL of pre-cultures was diluted in 200 mL TB containing 50 μg mL−1 kanamycin. Then they were incubated to reach an optimum density (OD) between 0.8 and 1.0 at 600 nm wavelength. The cells were induced by a final concentration of 0.5 molar IPTG for 2 hours and then were sedimented by means of centrifugation. The cell lysis was done by sonication and a Ni-NTA column was used to elute the attached proteins. The purity of the recombinant protein was evaluated by 16% Tris-Tricine.21 Since brazzein has no tryptophan in its structure, the absorbance was measured as the ratio of the 205/208 wavelength to determine the concentration of the proteins.1Elimination of the His-tag tail and purification of recombinant proteins
Cyanogen bromide was used to eliminate His-tag from the recombinant protein. Approximately 30–50 mg cyanogen bromide was added to 8 mL of protein solution which was prepared to 0.2 mg mL−1 concentration, pH 1.5–1.7, for a final concentration of 0.1 mmol. The samples were incubated in darkness at 25 °C for 18 hours.22 Buffer exchange was then performed with a dialysis buffer of 89 mmol and pH 7.6, using an Amicon-ultra 3 kDa column. The separation of eliminated proteins from the un-eliminated ones was performed by means of a Ni-NTA column. Desalination and condensation of the samples were conducted by a dialysis buffer and Amicon-ultra-0.5 mL 3 kDa. The quality of purification was investigated by 16% Tris-Tricine gel.Taste test
The tests for the solutions of the WT and mutants with defined concentrations were carried out according to the procedure of Lee et al.23 1% sucrose solution was also exploited for comparison. For the taste test, 5 men and 6 women, aged 20–39 with a normal sense of taste, were selected for taste evaluation. 200 μL of samples with the desired concentration was delivered to the apex of the tongue by a pipette. After every test, the mouth was washed with a cup of tap water. Then its sweetness was reported on a scale of basic weight in comparison with sucrose’s sweetness.13In the taste test experiment, for comparison of the sweetness power of a protein relative to sucrose, the lowest quantity of sucrose essential for sensing the sweetness (1 gram per mL) is divided by the lowest quantity of protein for similar sensing. The resulting parameters are reported as g g−1 and may be converted to the minimum number of protein molecules for sweetness sensing using the following equation:
| Number of molecules = (gram sucrose/gram protein) × (molecular mass of protein/molecular mass of sucrose) |
For preventing any experimental bias, in performing the taste test assay for protein variants, all participants as well as experiment conductors were not aware of the type of protein (WT or mutants) and the activity of the protein variants was assayed using a double-blind procedure.
Structural studies
Urea denaturation curves were then fitted to eqn (1) with the kaleidagraph analysis software.24
 | (1) |
The free energy of unfolding in the presence of urea, ΔG([urea]), is related to the concentration of denaturants as eqn (2):25
| ΔG([urea]) = ΔG([H2O]) − m([urea]) | (2) |
Accordingly, the free energy of unfolding in the absence of urea, ΔG([H2O]), is:
| ΔG([H2O]) = m([urea])50% | (3) |
Results and discussion
Bioinformatics studies
Among 20 different amino acids, four of them are considered as charged residues including arginine (R) and lysine (K) as positively charged residues as well as aspartic acid (D) and glutamic acid (E) as negatively charged residues. Hence, for changing the charge of the ninth position from negative to positive, E should be replaced with K and/or R. However, in order to evaluate the effect of charge on the structure and function of the protein, other structural properties of the new residue should be similar to the original residue except that of the charge. According to this argument the side chain of K is more similar to E in comparison with R. So the E9K mutant was the first candidate for our experimental design. For evaluating the structural and functional effects of local flexibility, Gly with a high degree of conformational space was selected as the other residue for replacing with E9.The structures of the mutants were modelled by MODELLER program version 9v7 using the crystal structure of WT brazzein (PDB: 4HE7) as a model (Fig. 1).
 | ||
| Fig. 1 Graphical description of mutations. Lower panel: the ribbon diagram of the whole structure of brazzein. The site of mutation is shown by a white arrow. Upper panel: representation of the accessibility of residues at the site of mutation for the WT and mutant proteins. The structure of the WT protein was obtained from the protein data bank (PDB code: 4HE7). The three-dimensional structure of the mutants was constructed with MODELLER program (version 9v7) using the primary sequences of protein variants in PIR format as input for the program and the structure of the WT protein was used as a template. | ||
The results of the Ca root-mean-square deviation (RMSD) value between the WT and mutants and other structural evaluation scores, calculated by Verify3D, Errat and ProCheck, showed the reliability of the structural models which were selected for further studies (Table 1).
The changes in the accessibility of residues in the WT and mutants were calculated by the VADAR server and are depicted in Fig. 2. It appears that internalization of Val7 as a hydrophobic residue accompanied by more accessibility of Lys9 in the E9K mutant may help the protein to gain more stability relative to the WT protein. More exposure of Val7 in the E9G variants may result from more flexibility of the protein as a consequence of replacing Glu9 by Gly. It was also found that Lys6 becomes more exposed to water molecules in the E9G and E9K mutants compared to in the WT protein.
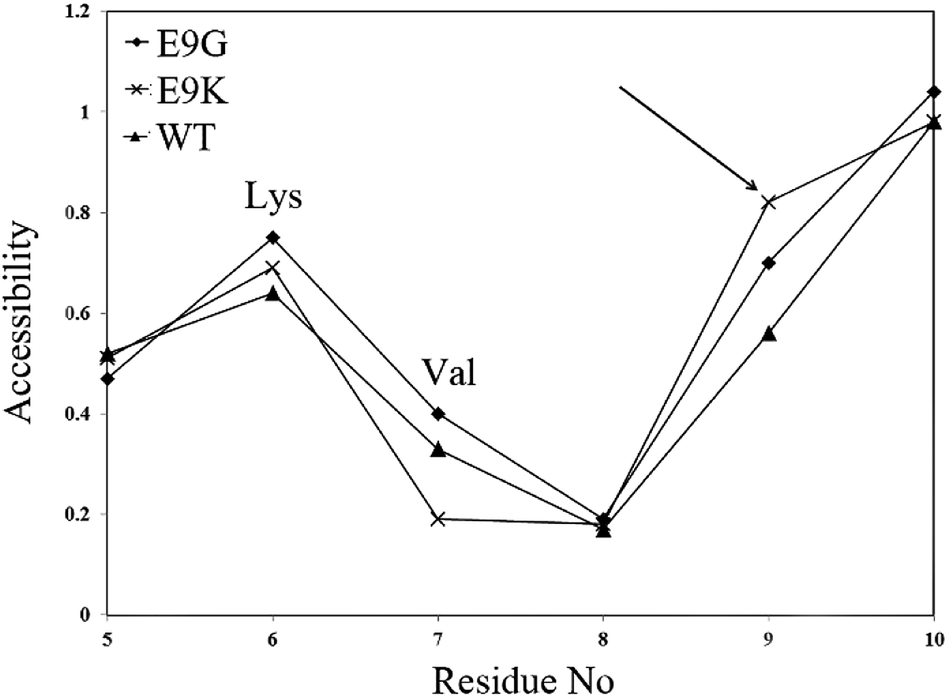 | ||
| Fig. 2 The output of the VADAR program indicating the alterations in the accessibility of residues upon mutation. The site of mutation is shown by the arrow. The crystal structure of the WT protein and structural models of the mutants were used as input for running the program. | ||
It can be seen that selection of the position of mutation was based on other reports concerning the effects of size and charge on the sweetness power of brazzein.5,10,14 In addition, physicochemical assumptions including size and conformational space as well as the accessibility of position 9 and the ability of new residues for interaction with polar solvent residues were also considered.
Site-directed mutagenesis and sequencing
To understand the structural and functional role of Glu9 in brazzein, two mutations including E9K and E9G were constructed by site-directed mutagenesis. Analysis of the PCR products using 1% agarose electrophoresis gel upon the quick chain site-directed procedure shows an amplification band with a relative length of 5560 bp which is equal to producing brazzein of 88 amino acids in length (in addition to His-tag) (data not show). Following sequencing of the PCR products, the validity of mutation and framework reading for recombinant brazzein were confirmed.Expression and purification of recombinant brazzein
E. coli strain SHuffle® T7 Express system is a good choice appropriate for increasing the capacity for correct folding of proteins containing disulfide bonds.26Examining the expression pattern of recombinant brazzein by Tris-Tricine gel showed a single band in the 10 kDa region revealing the expression of recombinant brazzein with His-tag (Fig. 3). Upon elimination of His-tag by cyanogen bromide the recombinant protein was observed as a single and pure band in the 6.5 kDa region (Fig. 3).
 | ||
| Fig. 3 The results of electrophoresis by 16% Tris-Tricine gel for native brazzein and its mutants in different stages of expression and purification in E. coli strain SHuffle® T7 Express: (1) the supernatant of lysate extract of bacteria after induction, (2) the supernatant of lysate extract of bacteria before induction, (4, 5 and 6) the WT, E9G and E8K mutants with His-tag after purification by Ni-NTA, (7, 8 and 9) the WT and mutant samples after histidine tag elimination, and (M) protein molecular weight marker with a size of 3.4–100 kDa (Thermo scientific protein ladder). | ||
Sweetness assessment
It was found that sweetness is generally dependent on the formation of hydrogen bonds between sweet molecules and sweet taste receptors.14 The cysteine-rich domain of T1R3 is responsible for the sweet taste of brazzein, thaumatin and monellin as sweet-tasting proteins. According to the Wedge model, in spite of the bulky size of sweet proteins relative to the three-dimensional structure of the pit, brazzein, thaumatin and monellin are bound to the open conformation of T1R2 or T1R3 receptors.27,28In the present study, two single mutations of brazzein at position 9 located at the first loop ranging from 9–19 were constructed and their sweetness was assessed and compared with that of the WT protein. Assessment of the sweetness threshold was carried out using 11 case studies including 5 men and 6 women. The results of the taste panel test for the WT protein and its mutants are shown in Fig. 4 and Table 2. Evaluation of sweetness for the mutants and WT protein (Table 2 and Fig. 4) indicates that the substitution of Glu9 with lysine and glycine leads to a significant increase in the sweetness property. It may be concluded that mutation in the flexible loop of brazzein has a great impact on its sweetness which is in good agreement with previous reports on the functional properties of brazzein.14 Substitution of Glu by Gly in E9G variants results in decreasing the negative charge, while the negative charge is converted to a positive charge in the E9K mutant. Furthermore, Gly has more conformational space and can increase the flexibility of the protein in the E9G mutant. It seems that decreasing the negative charge toward a positive one accompanied by increasing the flexibility of the protein is crucial for its interaction with the corresponding receptor and enhancement of the sweetness power of the protein.12,14,29
 | ||
| Fig. 4 The results of taste evaluation for WT and mutants. The results are gathered from 11 people, averaged and reported with corresponding errors. The black and hatched columns refer to the WT and mutant proteins, respectively. DW refers to distilled water. As shown in the figure, the sweetness power of the mutants increases in comparison with the WT protein. | ||
| Sweet-tasting molecule | Molecular mass (Da) | Experimental taste threshold | Sweetness in comparison to sucrose | ||
|---|---|---|---|---|---|
| (g (100 mL)−1) | μM | g g−1 | Molecule | ||
| Sucrose | 342.30 | 1 | 29![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
1 | 1 |
| WT | 6370.19 | 0.0004 | 0.6279 | 2510 | 46711 |
| E9K | 6369.25 | 0.0002 | 0.3140 | 5025 | 93501 |
| E9G | 6298.13 | 0.0002 | 0.3175 | 4980 | 91629 |
Structural studies
 | ||
| Fig. 5 Circular dichroism in the far-UV region for the WT and mutants. The far-UV CD spectra were recorded using a Jasco spectropolarimeter (JASCO J-810) using 0.2 mg mL−1 concentration of protein at pH 7.6 and at 25 °C. Data were recorded and analysed by Jasco analysis software. | ||
As we know, beta-strands are not stable alone. However, they could gain more stability by forming beta-sheets via intra-strand hydrogen bonds. Based on the geometry and pattern of hydrogen bonds, beta-sheets in globular proteins were also found to have a wide range of extended geometries with a range of flexibility and stability.
According to the CD spectra of Fig. 5, the E9K mutant with increased intensity in the CD spectrum has more organized, condensed and relatively stable β-sheets, whereas the E9G mutant shows more flexibility and distortion in its β-sheet content. Hence, the CD data spectra indicate that the secondary structure of the E9K mutant is more stable relative to the E9G and WT proteins.
The accessibility of hydrophobic patches at the surface of a protein may also be investigated by fluorescence spectroscopy using ANS as an extrinsic chromophore. The fluorescence spectra of the intrinsic and extrinsic chromophores of the WT and mutants are shown in Fig. 6a and b, respectively. It can be seen that there are two maxima in the intrinsic fluorescence emission of brazzein at 307 and 376 nm. The presence of two maxima in the fluorescence emission may be due to the presence of a resonance energy transfer mechanism on the structure of the protein and/or presence of two conformers in equilibrium. An exact structural examination of brazzein shows that Tyr39 and Phe38 may act as a donor–acceptor pair and the fluorescence energy of one residue can be absorbed by the electronic ground state of the other one for promotion to an excited state. This process is completed by fluorescence emission of the acceptor at a higher wavelength near 376 nm. It is noticeable that the interaction of chromophores with each other may result in alteration in the energy levels of their ground and excited state. According to this hypothesis the first peak near 307 nm includes the fluorescence emission of Tyr residues at positions 8, 11, 24, 51 and 54 and the other peak at 376 nm originates from the interaction of two remaining chromophores at positions 38 and 39.
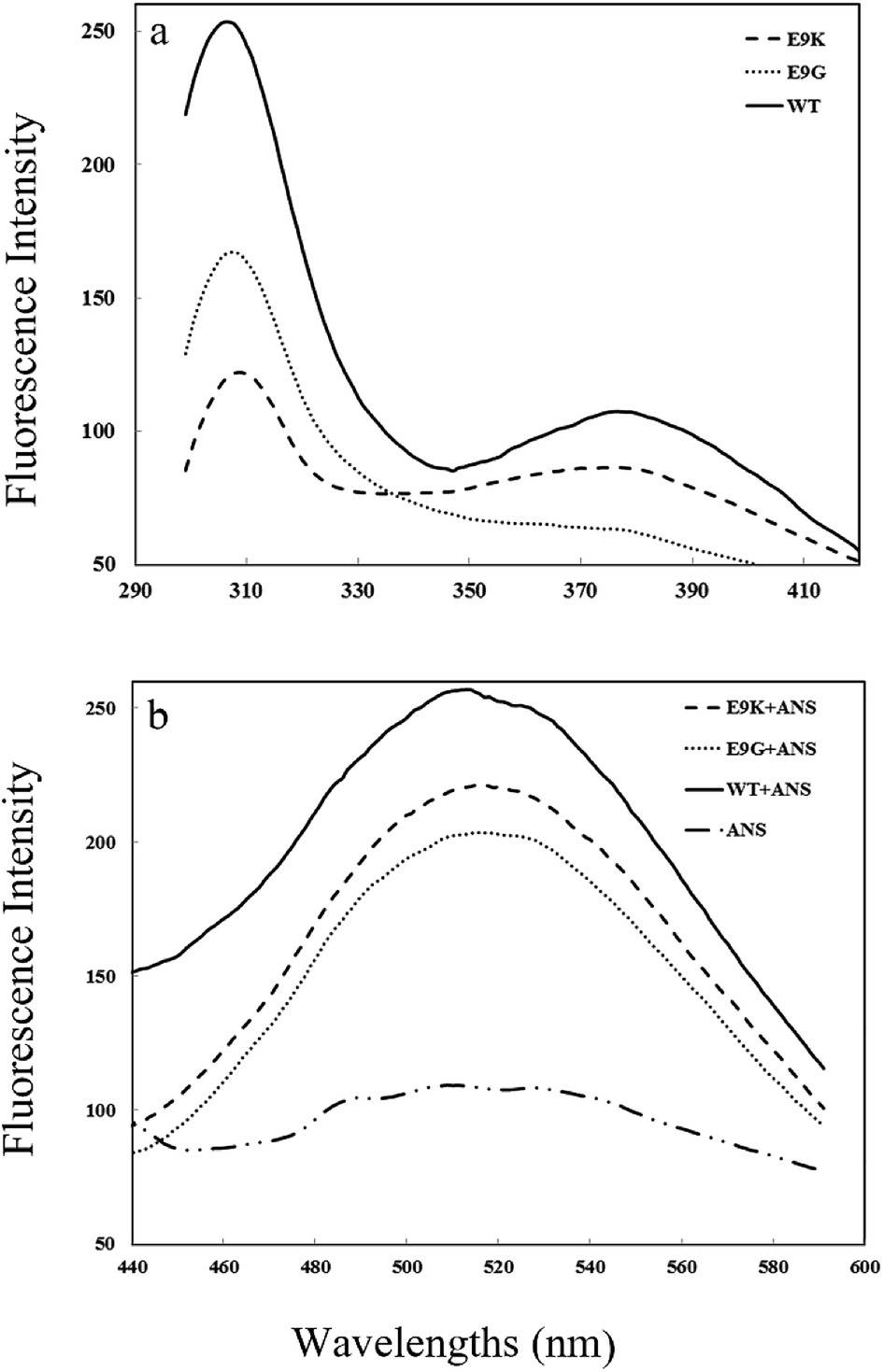 | ||
| Fig. 6 Intrinsic (a) and extrinsic (b) emission spectra for the WT and mutants. The spectra were recorded at 25 °C and at pH 7.6 using a Cary Eclipse spectrophotometer. The excitation wavelength was 280 and 370 nm for intrinsic and ANS fluorescence, respectively. The emission spectra were recorded between 300 and 420 nm for intrinsic measurement and 440–600 nm for ANS-based fluorescence. | ||
However, more studies including construction of new single and double mutants particularly at positions 38 and 39 of brazzein are needed for investigating the fluorescence properties of brazzein.
It is worth mentioning that brazzein is involved in internal quenching in its intrinsic fluorescence mechanism, so that its fluorescence emission increases with increasing the concentration of urea during the unfolding process (as is seen in the flowing reports on thermodynamics data). The internal quenching of fluorescence emission may be attributed to the vicinity of disulfide bonds to the chromophores which are involved in fluorescence emission. Investigation of the interaction of brazzein using a PIC server indicates that there are 4 disulfide bonds in its structure including C16–C37, C22–C47, C16–C37, and C4–C52. The main chains of Tyr8 and Tyr51 interact with Cys47 and Cys4, respectively. The aromatic part of Tyr8 may also interact with Cys47 as well as Cys49 and that of Tyr11 interacts with Cys16 and Cys47. Based on these assumptions it may be concluded that a part of the quenching effect originates from absorption of emitted photons of tyrosine residues, especially by Tyr8, Tyr51 and Tyr11. On the other hand, negatively charged residues may act as quenchers in the vicinity of chromophores. The main chains of Tyr11 and Tyr24 are spatially located near the main chains of Glu9 and Glu20, respectively. Additionally, the side chains of Tyr8 and Tyr11 have established hydrogen bonds with the side chains of Asp29 and Asp25, respectively. Accordingly, it may be concluded that Tyr8, Tyr11 and Tyr24 are involved in the suggested quenching.
Hence, the fluorescence intensity at 307 nm in Fig. 6a indicates that the tertiary structure of the protein around the tyrosine residues is more compact in the E9K mutant, while tyrosine residues are more exposed to a polar environment in the WT and E9G mutant. According to the ANS fluorescence data in Fig. 6b, the maximum fluorescence intensity of the WT protein is greater than that of the E9K and E9G mutants. Increasing the ANS fluorescence intensity in the WT protein is a consequence of increasing the number of photons emitted from the protein which in turn can be attributed to the increase of the number of surface hydrophobic patches of the WT protein accessible to ANS molecules. The absence of a wavelength shift in ANS fluorescence indicates that the binding strength of the ANS molecules to hydrophobic patches at the surface of the protein is not changed upon mutation.
These data together indicate that overall the tertiary structure of the E9G and E9K mutants is more compact in comparison to the WT protein and the local tertiary structure around tyrosine residues in the WT and E9G mutant is more exposed to a polar environment.
 | ||
| Fig. 7 Intrinsic fluorescence spectra of the WT protein measured at different concentrations of urea. At first a fixed volume of protein stock solution was mixed with defined volumes of buffer solution and fresh urea stock solution (10 M). 10 μL of DTT was also added to all the reaction mixture for reducing disulfide bonds. Upon 8 hours of incubation, fluorescence emission of all samples was measured between 300 and 420 nm with excitation wavelengths of 280 nm at 25 °C and at pH 7.6 using a 5 nm slit for both excitation and emission monochromators. | ||
Examination of different wavelengths in Fig. 7 indicates that the fluorescence intensity at 380 nm as a function of urea concentration has a mathematical behaviour as a sigmoid-like curve. The scatter-grams of these curves for all protein variants known as isothermal urea denaturation curves are shown in Fig. 8. The continuous lines represent the best fit of experimental data to eqn (1) describing the unfolding reaction of the WT and mutants assuming a two-state model.
 | ||
| Fig. 8 Modelling of experimental equilibrium denaturation data. Experimental data of denaturation experiments were fitted to eqn (1) by kaleidagraph analysis software. WT (Δ), E9K (×) and E9G (□). The resulting data of thermodynamics studies are provided in Table 3. | ||
The outputs of modelling the experimental data to eqn (1) are provided in Table 3.
| Protein variants | ΔG(H2O)a | m-Valuea | [Urea]50% |
|---|---|---|---|
| a Standard deviations are calculated based on three or four replicates of experiments.b m-Value in kcal mol−2, [urea]50% in M, and ΔG(H2O) in kcal mol−1. | |||
| WT | 1.97 ± 0.09 | 0.52 ± 0.02 | 3.79 ± 0.22 |
| E9G | 1.81 ± 0.03 | 0.56 ± 0.04 | 3.23 ± 0.24 |
| E9K | 2.53 ± 0.07 | 0.55 ± 0.09 | 4.60 ± 0.76 |
Since the structural integrity and conformational stability of proteins are mainly related to intramolecular weak interactions such as hydrogen bonds, hydrophobic, van der Waals and electrostatic interactions as well as the intermolecular interactions of surface exposed residues with polar solvent molecules, substitution of each amino acid by another one may result in a small change of interaction energy. Accordingly, the magnitude of ΔG(H2O) in Table 3 is biologically significant. However, T-test analysis of the resulting values of ΔG(H2O) indicates that the differences between means are statistically significant at p < 0.05.
According to the thermodynamics data of Table 3, ΔG(H2O) of the E9K mutant is greater than that for the WT and E9G mutants indicating more stability of the tertiary structure of the E9K mutant compared to the WT and E9G one. The thermodynamic m-value is a parameter which is related to the difference in accessible surface area between the folded and unfolded state of the protein. A similar magnitude of m-values for all protein variants indicates that the greater value of ΔG(H2O) for the E9K mutant is more related to [urea]50% rather than the m-value. According to the hydropathy index of Kyte and Doolittle, Lys with a hydropathy score of −3.9 has thermodynamically more tendency for interaction with water molecules relative to Glu and Gly with scores of −3.5 and −0.4, respectively.30 Based on these findings the greater stability of the E9K mutant originates mainly from more favourable interaction of Lys9 at the surface of the E9K mutant with a polar environment. By similar reasoning and according to Fig. 1, the mutation leads to more internalization of Val7 in the E9K mutant which can help the protein to gain more stability.
Conclusions
In conclusion, it can be deduced that brazzein’s alkalinity at the surface of a protein has a significant role on its sweetness, so that changing its net charge toward a positive one can influence its interaction with the sweet taste receptor. Our data indicate that brazzein has interesting fluorescence properties concerning resonance energy transfer and internal quenching effects which may be confirmed using convenient single and double mutations involving residues Phe38 and Tyr39.Acknowledgements
Financial support of this work was provided by Iran National Science Foundation (Fund Number: 90007290). We would also like to thank the Research Council of the University of Zanjan. We also appreciate technical support from Institute of Biochemistry and Biophysics (IBB) of the university of Tehran and Institute for Advanced Studies in Basic Sciences of Zanjan regarding circular dichroism and fluorescence measurements. The authors have declared no conflict of interest.References
- F. M. Assadi-Porter, S. Patry and J. L. Markley, Protein Expression Purif., 2008, 58, 263–268 CrossRef CAS PubMed.
- F. Mansouri, J. Sci., Islamic Repub. Iran, 2011, 22, 105–110 CAS.
- R. Kant, Nutr. J., 2005, 4, 1 CrossRef PubMed.
- R. Wintjens, T. M. V. N. Viet, E. Mbosso and J. Huet, Plant Sci., 2011, 181, 347–354 CrossRef CAS PubMed.
- Z. Jin, V. Danilova, F. M. Assadi-Porter, D. J. Aceti, J. L. Markley and G. Hellekant, FEBS Lett., 2003, 544, 33–37 CrossRef CAS PubMed.
- Q. Zhao, J. Song, Z. Jin, V. Danilova, G. Hellekant and J. L. Markley, Biochemical and biophysical research communications, 2005, 335, 256–263 CrossRef CAS PubMed.
- P. Temussi, Cell. Mol. Life Sci., 2006, 63, 1876–1888 CrossRef CAS PubMed.
- K.-I. Nakajima, K. Yokoyama, T. Koizumi, A. Koizumi, T. Asakura, T. Terada, K. Masuda, K. Ito, A. Shimizu-Ibuka and T. Misaka, PLoS One, 2011, 6, e19448 CrossRef CAS PubMed.
- F. M. Assadi-Porter, F. Abildgaard, H. Blad, C. C. Cornilescu and J. L. Markley, Chem. Senses, 2005, 30, i90–i91 CrossRef CAS PubMed.
- J.-W. Lee, J.-E. Cha, H.-J. Jo and K.-H. Kong, Food Chem., 2013, 138, 1370–1373 CrossRef CAS PubMed.
- C. C. Cornilescu, G. Cornilescu, H. Rao, S. F. Porter, M. Tonelli, M. L. DeRider, J. L. Markley and F. M. Assadi-Porter, Proteins: Struct., Funct., Bioinf., 2013, 81, 919–925 CrossRef CAS PubMed.
- F. M. Assadi-Porter, E. L. Maillet, J. T. Radek, J. Quijada, J. L. Markley and M. Max, J. Mol. Biol., 2010, 398, 584–599 CrossRef CAS PubMed.
- F. M. Assadi-Porter, D. J. Aceti and J. L. Markley, Arch. Biochem. Biophys., 2000, 376, 259–265 CrossRef CAS PubMed.
- S.-Y. Yoon, J.-N. Kong, D.-H. Jo and K.-H. Kong, Food Chem., 2011, 129, 1327–1330 CrossRef CAS.
- F. M. Assadi-Porter, F. Abildgaard, H. Blad and J. L. Markley, J. Biol. Chem., 2003, 278, 31331–31339 CrossRef CAS PubMed.
- D. E. Walters and G. Hellekant, J. Agric. Food Chem., 2006, 54, 10129–10133 CrossRef CAS PubMed.
- M. A. Martí-Renom, A. C. Stuart, A. Fiser, R. Sánchez, F. Melo and A. Šali, Annu. Rev. Biophys. Biomol. Struct., 2000, 29, 291–325 CrossRef PubMed.
- K. G. Tina, R. Bhadra and N. Srinivasan, Nucleic Acids Res., 2007, 35, W473–W476 CrossRef CAS PubMed.
- N. Guex and M. C. Peitsch, Electrophoresis, 1997, 18, 2714–2723 CrossRef CAS PubMed.
- L. Willard, A. Ranjan, H. Zhang, H. Monzavi, R. F. Boyko, B. D. Sykes and D. S. Wishart, Nucleic Acids Res., 2003, 31, 3316–3319 CrossRef CAS PubMed.
- H. Schägger, Nat. Protoc., 2006, 1, 16–22 CrossRef PubMed.
- F. M. Assadi-Porter, D. J. Aceti, H. Cheng and J. L. Markley, Arch. Biochem. Biophys., 2000, 376, 252–258 CrossRef CAS PubMed.
- J.-J. Lee, J.-N. Kong, H.-D. Do, D.-H. Jo and K.-H. Kong, Bull. Korean Chem. Soc., 2010, 31, 3830–3833 CrossRef CAS.
- M. M. Santoro and D. Bolen, Biochemistry, 1988, 27, 8063–8068 CrossRef CAS PubMed.
- N. C. Pace and K. L. Shaw, Proteins: Struct., Funct., Genet., 2000, 4, 1–7 CrossRef.
- J. Lobstein, C. A. Emrich, C. Jeans, M. Faulkner, P. Riggs and M. Berkmen, Microb. Cell Fact., 2012, 11, 56 CrossRef CAS PubMed.
- P. A. Temussi, FEBS letters, 2002, 526, 1–4 CrossRef CAS PubMed.
- R. Spadaccini, F. Trabucco, G. Saviano, D. Picone, O. Crescenzi, T. Tancredi and P. A. Temussi, J. Mol. Biol., 2003, 328, 683–692 CrossRef CAS PubMed.
- H.-D. Do, H.-J. Jo, D.-H. Jo and K.-H. Kong, Bull. Korean Chem. Soc., 2011, 32, 4106–4108 CrossRef CAS.
- J. Kyte and R. F. Doolittle, J. Mol. Biol., 1982, 157, 105–132 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |