
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExhaustive 3D-QSAR analyses as a computational tool to explore the potency and selectivity profiles of thieno[3,2-d]pyrimidin-4(3H)-one derivatives as PDE7 inhibitors†
Elena
Cichero
*,
Chiara
Brullo
,
Olga
Bruno
and
Paola
Fossa
Department of Pharmacy, Section of Medicinal Chemistry, School of Medical and Pharmaceutical Sciences, University of Genoa, Viale Benedetto XV n. 3, 16132, Genoa, Italy. E-mail: cichero@unige.it; Fax: +39-0103538357; Tel: +39-0103538370
First published on 21st June 2016
Abstract
The development of selective ligands binding to specific PDE isoforms represents an urgent need in medicinal chemistry, being a necessary strategy to identify many more drug-like compounds, to be investigated for several therapies. Concerning inflammation, rational design of selective PDE7 inhibitors over PDE4 could lead to derivatives endowed with a better safety profile, showing limited side-effects. In this context, thieno[3,2-d]pyrimidin-4(3H)-one-based compounds have been recently studied as a series of potent phosphodiesterase type 7 (PDE7) inhibitors, most of them being selective over other PDE enzymes, such as PDE4B. This work describes a computational study based on docking calculations followed by Comparative Molecular Fields Analysis (CoMFA) and Comparative Molecular Similarity Indices Analysis (CoMSIA), in order to better elucidate the pharmacophore features of this series of PDE7 inhibitors. The results reveal the ligand-based approach as a promising strategy to better investigate the potency and selectivity issues of PDE7 inhibitors. In addition, the results also allowed robust statistical models able to predict the potency and selectivity trend of new analogues prior to synthesis to be obtained.
Introduction
Phosphodiesterases (PDEs) represent a complex superfamily of enzymes that decrease c-AMP and c-GMP levels by hydrolysing them to their inactive 5′-monophosphate metabolites.1PDEs have been classified into 11 families based on sequence similarity, inhibitor sensitivity, and biochemical properties.2
The inhibition of tissue-specific phosphodiesterases has been shown to be an effective therapeutic approach for several diseases. PDE3 inhibitors are useful for heart diseases,3,4 being this enzyme particularly abundant in cardiac muscle and vascular smooth muscle.5 The inhibition of PDE5 is involved in the development of drugs for the treatment of erectile dysfunction.6
PDE4 is expressed predominantly in proinflammatory and immune cells, therefore the related inhibitors have been studied extensively in attempts to find ways to control the inflammatory conditions associated with asthma and chronic obstructive pulmonary disease.7
Although the effectiveness of PDE4 inhibitors for the treatment of airway inflammation is highly supported,8 the clinical use of PDE4 inhibitors seems to be restricted by the presence of prominent side effects, like nausea, emesis and sedation. In this context, recent studies performed around a series of PDE4D inhibitors acting without inducing emesis, suggested that the undesired effects associated with previously developed inhibitors may be probably due to their non-selective action.9
On these basis, an appealing strategy to design new more safe therapeutic agents for inflammation may be to target other phosphodiesterase isoenzymes that are specifically expressed in proinflammatory and immune cells, relying on much more selective compounds.
PDE7 is a cAMP-specific phosphodiesterase and consists of two genes (PDE7A and PDE7B).10–13
In particular, in humans and mice, the immune system (thymus, spleen, lymph nodes, and blood leukocytes)14 is a rich source of PDE7A. On these basis, PDE7 is considered to be a possible target for treating various diseases, including leukemias, central nervous system disorders and airway diseases.15–17
Up to now, several PDE7 inhibitors (PDE7Is) have been discovered and reported in literature18–22 and, among them, has been proposed a series of thieno[3,2-d]pyrimidin-4(3H)-one derivatives, endowed with promising potency and selectivity profiles within the PDE7A isoform.20,22
In terms of rational drug design process, with the aim at exploring the inhibitor structural features turning in potency and selectivity variations, the computational approach could rely on structure-based and ligand-based strategies. Unfortunately, due to the very similarity trend observed among all the PDE catalytic sites, the ligand-based approach, rather than the structure-based, could represent a much more promising tool to deeply investigate the selectivity issue around PDEIs, as we previously discussed around the PDE4DIs.23
In this work we applied docking-based Comparative Molecular Fields Analysis (CoMFA) and Comparative Molecular Similarity Indices Analysis (CoMSIA), starting from the X-ray crystallographic data about PDE7A in complex with the thieno[3,2-d]pyrimidin-4(3H)-one derivative 56 (Fig. 1). In details, we developed two series of 3D-QSAR analyses (namely model A and B) exploring through quantitative methods the main features responsible of PDE7Is potency and selectivity profiles, respectively.
 | ||
| Fig. 1 Chemical structures of compound 56 and of the related analogues. | ||
The results allowed us to derive robust statistical models to be used to predict the potency and selectivity trend of new analogues prior to synthesis and also to pave the way for the further design of more effective ligands.
Results and discussion
Docking-based ligand alignment
Docking calculations here proposed were applied with the aim to derive a reliable alignment of compounds 1–72 (Table 1), selected from literature,20–22 to be used for the following CoMFA and CoMSIA studies. Notably, the development of docking-based 3D-QSAR studies in most cases proved to be particularly effective and more reliable than those applied on standard conformational analysis-based approaches, as described in literature.24–28|
|
||||||
|---|---|---|---|---|---|---|
| Comp. | R | R1 | R2 | R3 | PDE7 pIC50 | PDE4B pIC50 |
| 1 | –CH3 |
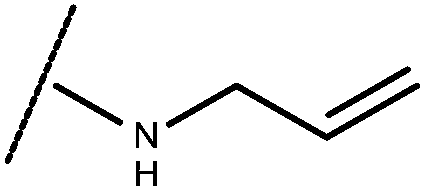
|
–H |

|
6.80 | — |
| 2 | –CH3 |

|
–H |

|
6.96 | — |
| 3 | –CH3 |

|
–H |

|
6.51 | — |
| 4 | –CH3 |

|
–H |

|
5.17 | — |
| 5 | –CH3 |

|
–H | –H | 5.30 | — |
| 6 | –CH2CH3 |

|
–H | –H | 6.82 | 5.31 |
| 7 | –CH2CH3 |

|
–H | –H | 6.13 | — |
| 8 | –CH2CH3 |
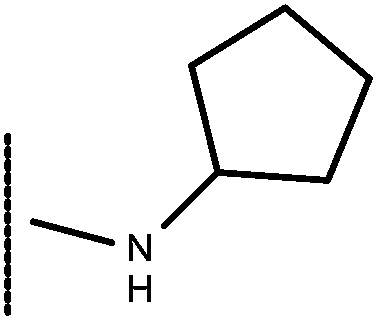
|
–H | –CH3 | 5.96 | — |
| 9 | –CH2CH3 |

|
–H | –CH2CH3 | 5.19 | 5.46 |
| 10 | –CH2CH3 |

|
–H |

|
5.59 | — |
| 11 | –CH2CH3 |

|
–CH3 | –H | 7.01 | 5.42 |
| 12 | –CH2CH3 |

|
–CH2CH3 | –H | 7.38 | 5.23 |
| 13 | –CH2CH3 |
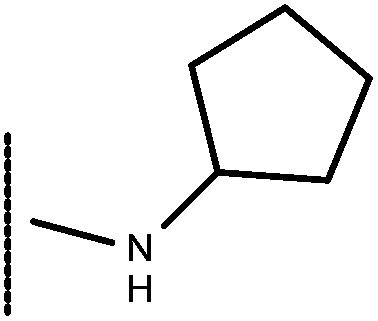
|

|
–H | 7.52 | 5.43 |
| 14 | –CH2CH3 |
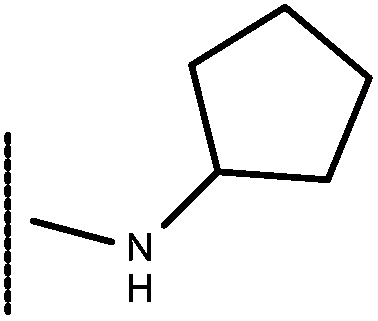
|

|
–H | 7.35 | 5.52 |
| 15 | –CH2CH3 |

|

|
–H | 7.17 | 5.49 |
| 16 | –CH2CH3 |
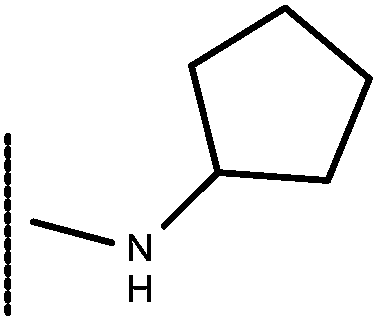
|

|
–H | 8.38 | 5.85 |
| 17 | –CH2CH3 |

|

|
–H | 7.72 | 6.09 |
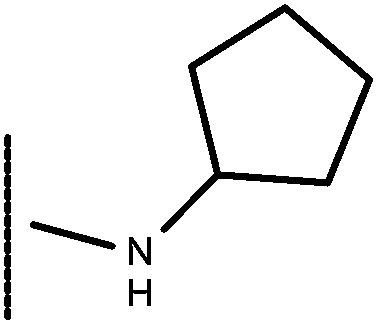
| 18 | –CH2CH3 |

|
|
–H | 8.14 | 5.96 |
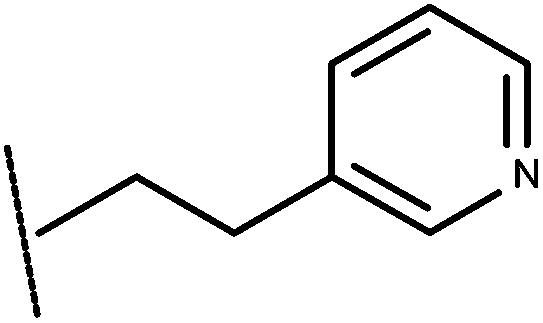
| 19 | –CH2CH3 |
|

|
–H | 7.41 | 5.47 |
| 20 | –CH2CH3 |

|

|
–H | 8.25 | 6.14 |
| 21 | –CH2CH3 |

|
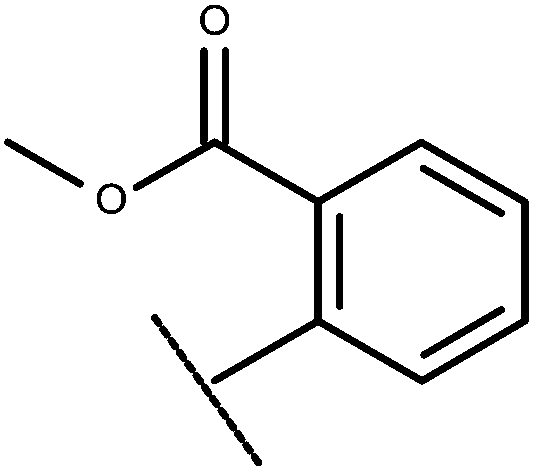
|
–H | 6.42 | 5.42 |
| 22 | –CH2CH3 |

|

|
–H | 8.30 | 6.32 |
| 23 | –CH2CH3 |

|

|
–H | 8.31 | 6.00 |
| 24 | –CH2CH3 |

|
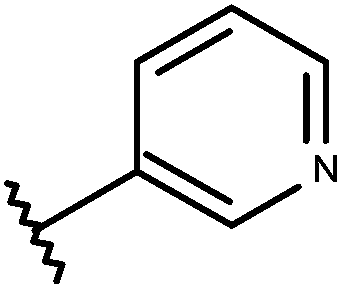
|
–H | 8.70 | 6.42 |
| 25 | –CH2CH3 |

|
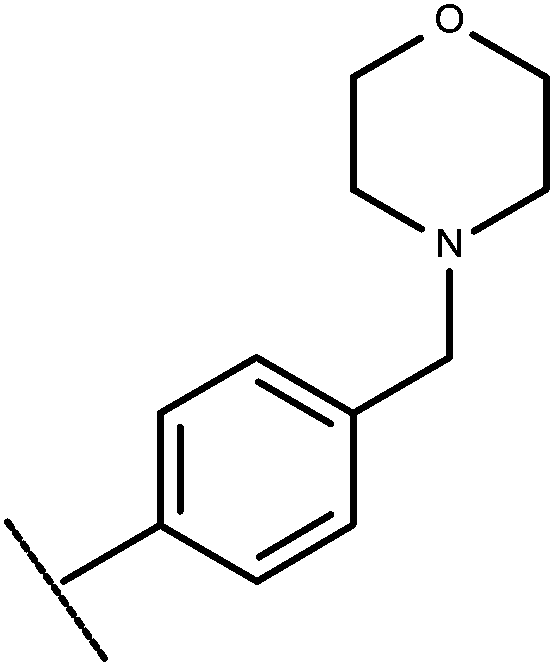
|
–H | 8.64 | 6.19 |
| 26 | –CH2CH3 |

|
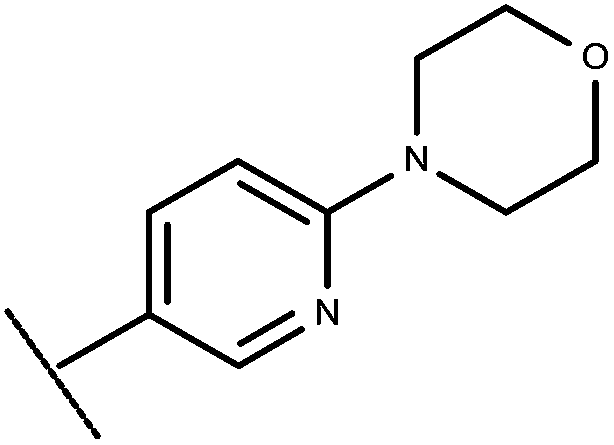
|
–H | 9.00 | 6.28 |
| 27 | –CH2CH3 |
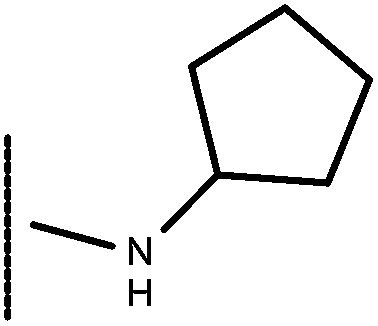
|
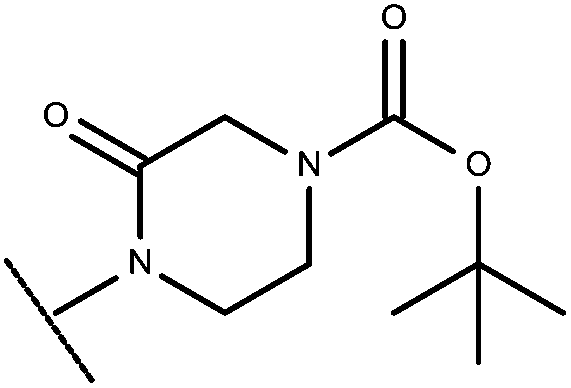
|
–H | 7.92 | 5.48 |
| 28 | –CH2CH3 |

|
–H | –H | 6.54 | 5.13 |
| 29 | –CH2CH3 |

|
–H | –H | 7.09 | 5.37 |
| 30 | –CH2CH3 |
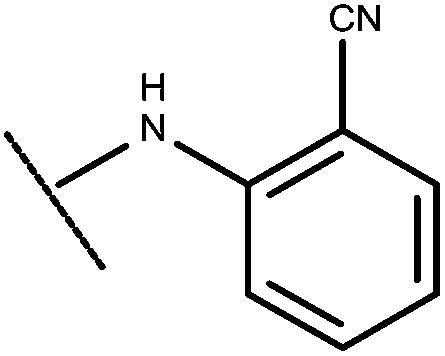
|
–H | –H | 5.64 | — |
| 31 | –CH2CH3 |

|
–H | –H | 7.21 | 5.57 |
| 32 | –CH2CH3 |
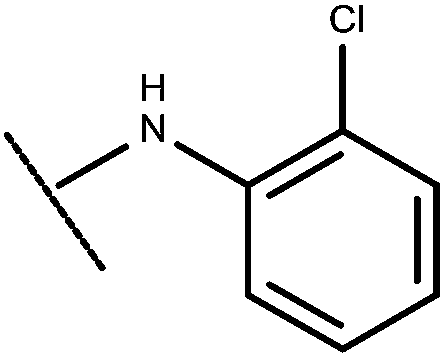
|
–H | –H | 7.39 | 5.52 |
| 33 | –CH2CH3 |

|
–H | –H | 6.20 | 7.52 |
| 34 | –CH2CH3 |

|
–H | –H | 7.22 | 5.28 |
| 35 | –CH2CH3 |

|

|
–H | 7.60 | 6.17 |
| 36 | –CH2CH3 |
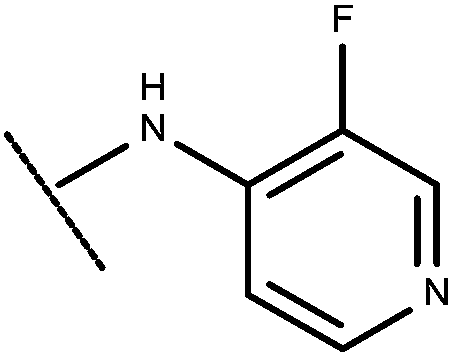
|

|
–H | 8.06 | 6.21 |
| 37 | –CH2CH3 |

|
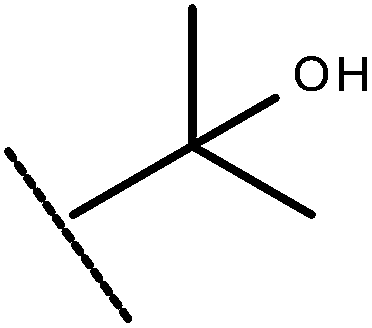
|
–H | 8.68 | 7.25 |
| 38 | –CH2CH3 |
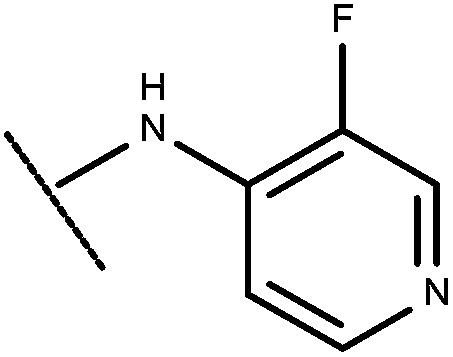
|

|
–H | 8.41 | 6.47 |
| 39 | –CH2CH3 |

|

|
–H | 8.96 | 6.51 |
| 40 | –CH2CH3 |

|
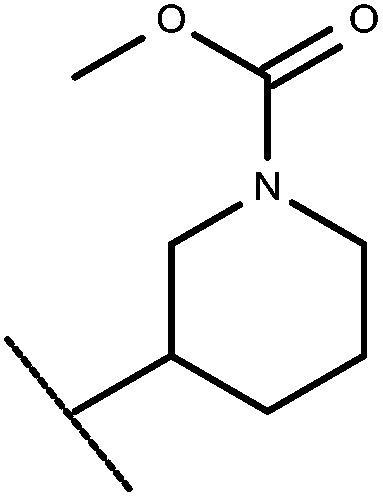
|
–H | 9.17 | 6.55 |
| 41 | –CH2CH3 |

|

|
–H | 8.92 | 6.74 |
| 42 | –CH2CH3 |

|
–H | –H | 9.49 | 7.70 |
| 43 | –CH2CH3 |

|
–H | –H | 5.54 | — |
| 44 | –CH2CH3 |
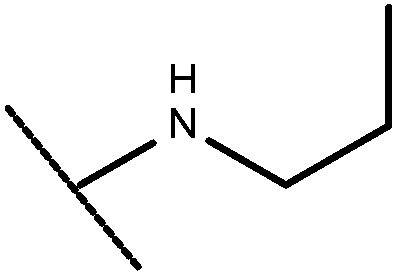
|
–H | –H | 5.85 | — |
| 45 | –CH2CH3 |

|
–H | –H | 6.43 | 7.96 |
| 46 | –CH2CH3 |

|
–H | –H | 5.33 | — |
| 47 | –CH2CH3 |

|
–H | –H | 5.82 | — |
| 48 | –CH2CH3 |

|
–H | –H | 5.59 | — |
| 49 | –CH2CH3 |

|
–H | –H | 6.52 | 5.32 |
| 50 | –CH2CH3 |

|
–H | –H | 6.82 | 5.31 |
| 51 | –CH2CH3 |

|
–H | –H | 6.13 | 5.28 |
| 52 | –CH2CH3 |

|
–H | –H | 6.37 | 7.92 |
| 53 | –CH2CH3 |

|
–H | –H | 5.64 | 7.62 |
| 54 | –CH2CH3 |

|
–H | –H | 5.68 | — |
| 55 | –CH2CH3 |
|
–H | –H | 7.22 | 5.28 |
| 56 | –CH2CH3 |

|

|
–H | 7.22 | 5.28 |
| 57 | –CH2CH3 |

|

|
–H | 7.22 | 5.28 |
| 58 | –CH2CH3 |

|

|
–H | 7.22 | 5.28 |
| 59 | –CH2CH3 |

|

|
–H | 8.34 | 6.38 |
| 60 | –CH2CH3 |
|
|
–H | 8.12 | 5.96 |
| 61 | –CH2CH3 |

|

|
–H | 6.72 | 4.85 |
| 62 | –CH2CH3 |

|
|
–H | 6.77 | 4.60 |
| 63 | –CH2CH3 |
|

|
–H | 8.17 | 6.36 |
| 64 | –CH2CH3 |

|

|
–H | 7.70 | 5.82 |
| 65 | –CH2CH3 |
|
|
–H | 8.29 | 6.25 |
| 66 | –CH2CH3 |

|

|
–H | 7.82 | 5.72 |
| 67 | –CH2CH3 |
|

|
–H | 8.08 | 5.85 |
| 68 | –CH2CH3 |

|
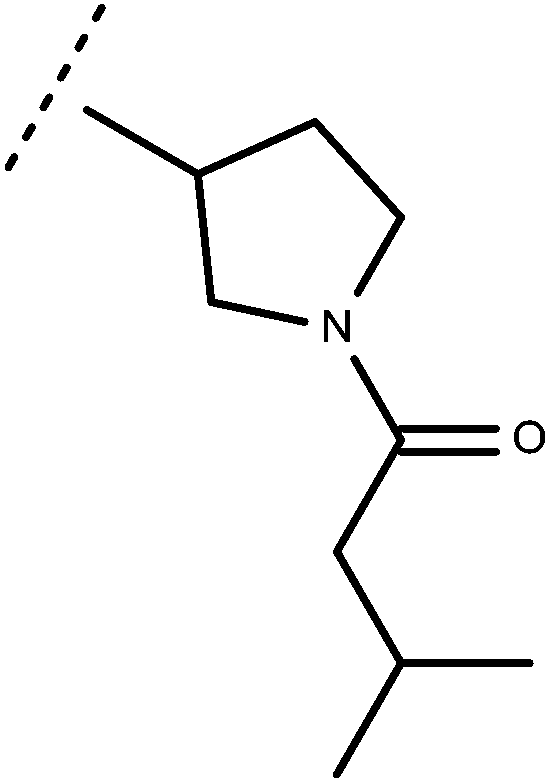
|
–H | 8.09 | 5.64 |
| 69 | –CH2CH3 |

|
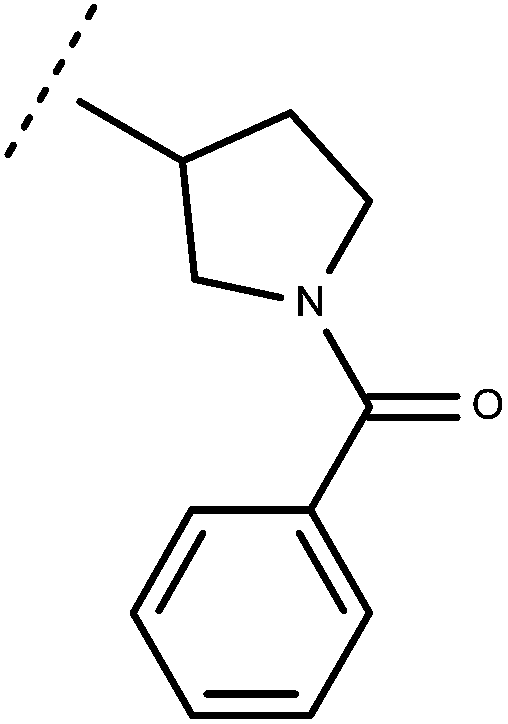
|
–H | 8.82 | 6.42 |
| 70 | –CH2CH3 |

|
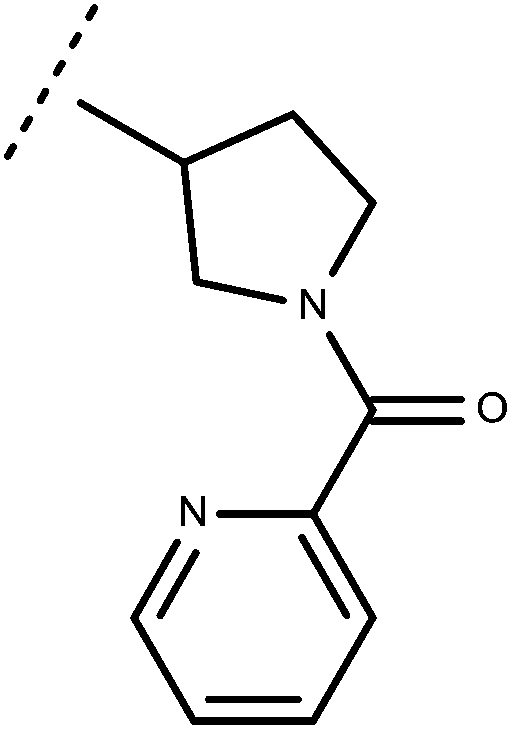
|
–H | 8.57 | 6.70 |
| 71 | –CH2CH3 |

|

|
–H | 8.85 | 6.85 |
| 72 | –CH2CH3 |

|

|
–H | 8.48 | 7.34 |

Thus, we performed our work using the available X-ray crystallographic data about compound 56, in complex with the PDE7A catalytic site (pdb code: 4Y2B).22
Based on the experimental data (see ESI S1†), the PDE7A inhibitor 56 displays one H-bond between the carboxamide oxygen atom and a glutamine residue (namely Gln413), that is highly conserved within all the PDE isoforms. In addition, the compound is stabilized at the enzyme catalytic site through water-bridges involving the aforementioned oxygen atom and Asn365, and also between the nitrogen atom of the propan-2-amine group and Tyr211 and Asp362.
The subsequent molecular docking calculations performed on the whole dataset allowed us to reasonably identified the most probable bioactive conformations of the thieno[3,2-d]pyrimidin-4(3H)-one derivatives here discussed.
First of all, as shown in Fig. 2a, we verified the reliability of the docking protocol through re-docking calculations on the reference compound 56, deriving a binding mode highly comparable with that disclosed by the X-ray data (4Y2B).
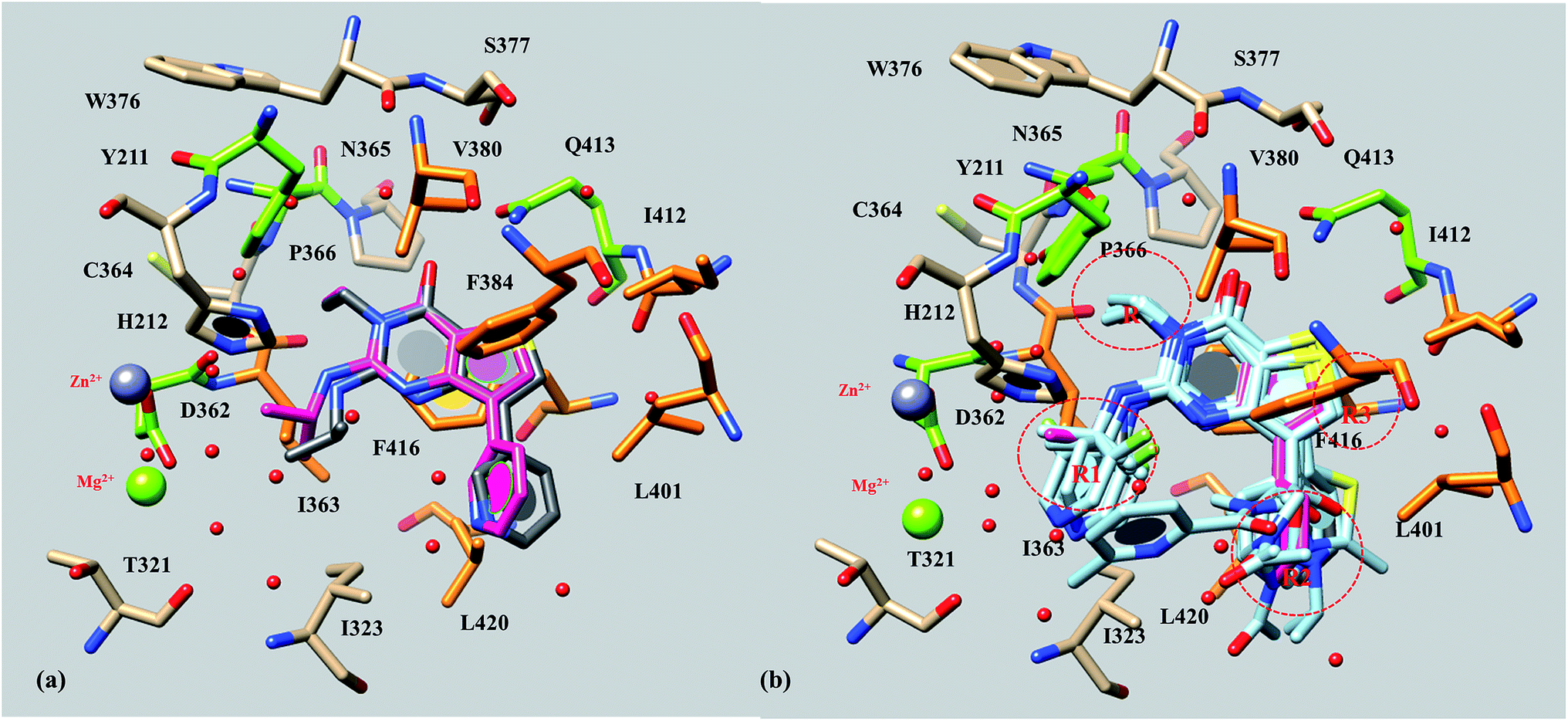 | ||
| Fig. 2 X-ray crystallographic conformation (C atom: grey) and docking pose (C atom: magenta) of 56 (a), and docking poses of compound 56 analogues (b) within the PDE7A catalytic site. Key polar and hydrophobic residues are highlighted in green and orange, respectively. The presence of R, R1, R2 and R3 substituents has been highlighted in (b) through dot circles. | ||
Concerning an overall analysis of docking results about the whole dataset, we can infer that the inhibitory ability of this series of molecules revolves around the establishment of H-bonds with Gln413 and by means of water-bridges through the carboxamide oxygen atom and the nitrogen one of the R1 substituent, as previously mentioned about 56.
In addition, the bicyclic core is in any case engaged in π–π stacking with Phe384 and Phe416, while the R, R2 and R3 substituents are projected towards Pro366; Ile323, Leu420; and Ile412, respectively (Fig. 2b). Notably, those derivatives bearing a proper substituent in R1 or bulky groups showing H-bonding moieties in R2, could be involved in further contacts with the Ile323 backbone, moving sometimes toward the metal binding pocket.
In order to derive some preliminary information about the putative role of thieno[3,2-d]pyrimidin-4(3H)-one derivatives as PDE4B ligands, we also explored the compounds binding mode at the catalytic site of this enzyme (pdb code: 3GWT).29 As shown in Fig. 3, the co-crystallized PDE4B quinolone inhibitor (PDE4B pIC50 = 8.4) was highly stabilized within the enzyme catalytic site through H-bond the conserved Gln443 and the nitrogen atom of the quinolone ring, and also a pattern of specific water-bridges revolved around the metal binding pocket. Conversely, compound 6 (PDE7 pIC50 = 6.82; PDE4B pIC50 = 5.31) was partially able to mimic these key contacts, detecting only one H-bond with the aforementioned glutamine residue through the oxygen atom of the carbonyl group. Notably, compound 6 displays a reversed arrangement within the PDE4B (if compared with the related one at the PDE7 cavity) being unable in this last case to be involved in water-mediated interactions at the metal binding pocket. As a consequence, the compound exhibits a poor potency profile towards PDE7.
 | ||
| Fig. 3 Docking poses of compound 6 (C atom; green) within the PDE4B catalytic site. The co-crystallized quinolone inhibitor shown and depicted in orchid. Most important residues are labelled. | ||
More in details, compound 6 oriented the R1 substituent towards Met431 and Ile450, while the overall thieno[3,2-d]pyrimidin-4(3H)-one scaffold was engaged in π–π stacking with Phe414 and Phe446. The bulky R2 and R3 groups of any related analogues proved to be exposed towards the metal binding pocket, being the R and R1 groups surrounded by a narrow lipophilic region.
Since for all compounds the best-docked geometries we derived within PDE7A proved to be in good agreement with the X-ray PDE7A/56 crystallographic data (for compound 56 itself by means of re-docking run, and also for the other analogues), all the compounds resulted to be already aligned and therefore submitted to the subsequent CoMFA30 and CoMSIA31 studies.
3D-QSAR analyses
CoMFA and CoMSIA analyses here reported were used to explore, through quantitative methods, the main features responsible of the thieno[3,2-d]pyrimidin-4(3H)-one derivatives potency and selectivity profiles to PDE7, over PDE4B (models A and B).For both the two models, CoMFA and CoMSIA calculations were developed using CoMFA steric and electrostatic fields, and CoMSIA steric, electrostatic, hydrophobic, H-bond acceptor and H-bond donor parameters, as independent variables. On the other hand, the inhibitor PDE7 pIC50 and the related “weighted” pIC50 were employed as dependent variables for model A and B, respectively (see Experimental section). For both the two models, compound pIC50 ranges covered at least four log orders of magnitude.
Concerning model A, CoMFA and CoMSIA analyses were performed by dividing compounds 1–72 into a training set (3–11, 13–20, 22, 23, 25–27, 29–34, 36–39, 41–52, 54–57, 59–63, 65–67, 69, 70, 72) for model generation and into a test set (1, 2, 12, 21, 24, 28, 35, 40, 53, 58, 64, 68, 71) for model validation. Model B CoMFA and CoMSIA studies were calculated including compounds 6, 11–13, 15–17, 19–24, 26, 28, 29, 31, 33, 37–40, 42, 45, 49–53, 56, 57, 60–64, 67, 68, 70–72 into the training set and choosing 14, 18, 25, 27, 32, 34–36, 41, 58, 59, 65, 66 and 69 for the test set.
The related model A and model B experimental and predicted pIC50 values are reported in Tables 2–5, while all statistical parameters supporting the two series of 3D-QSAR analyses are reported in Table 6 and detailed as follows.
| Compounds | Experimental pIC50 | CoMFA analysis | CoMSIA analysis | ||
|---|---|---|---|---|---|
| Predicted pIC50 | Residual | Predicted pIC50 | Residual | ||
| 3 | 6.51 | 6.30 | 0.21 | 6.45 | 0.06 |
| 4 | 5.17 | 4.92 | 0.25 | 4.66 | 0.51 |
| 5 | 5.30 | 5.50 | −0.20 | 5.78 | −0.48 |
| 6 | 6.82 | 6.40 | 0.42 | 6.28 | 0.54 |
| 7 | 6.13 | 6.29 | −0.16 | 6.34 | −0.21 |
| 8 | 5.96 | 5.92 | 0.04 | 5.82 | 0.14 |
| 9 | 5.19 | 5.49 | −0.30 | 5.56 | −0.37 |
| 10 | 5.59 | 5.40 | 0.19 | 5.81 | −0.22 |
| 11 | 7.01 | 7.14 | −0.13 | 6.79 | 0.22 |
| 13 | 7.52 | 7.51 | 0.01 | 7.23 | 0.29 |
| 14 | 7.35 | 7.51 | −0.16 | 6.96 | 0.39 |
| 15 | 7.17 | 7.10 | 0.07 | 7.32 | −0.15 |
| 16 | 8.38 | 8.47 | −0.09 | 8.24 | 0.14 |
| 17 | 7.72 | 7.84 | −0.12 | 7.72 | 0.00 |
| 18 | 8.14 | 8.32 | −0.18 | 8.24 | −0.10 |
| 19 | 7.41 | 7.37 | 0.04 | 7.39 | 0.02 |
| 20 | 8.25 | 8.35 | −0.10 | 8.24 | 0.01 |
| 22 | 8.30 | 8.42 | −0.12 | 8.58 | −0.28 |
| 23 | 8.31 | 8.34 | −0.03 | 8.49 | −0.18 |
| 25 | 8.64 | 8.81 | −0.17 | 8.59 | 0.05 |
| 26 | 9.00 | 8.76 | 0.24 | 8.90 | 0.10 |
| 27 | 7.92 | 8.03 | −0.11 | 8.09 | −0.17 |
| 29 | 7.06 | 7.01 | 0.05 | 7.04 | 0.02 |
| 30 | 5.64 | 6.04 | −0.40 | 6.41 | −0.77 |
| 31 | 7.21 | 6.87 | 0.35 | 7.00 | 0.21 |
| 32 | 7.39 | 6.91 | 0.48 | 7.25 | 0.14 |
| 33 | 6.20 | 6.38 | −0.18 | 6.34 | −0.14 |
| 34 | 7.22 | 6.86 | 0.36 | 6.72 | 0.50 |
| 36 | 8.06 | 8.19 | −0.13 | 8.27 | −0.21 |
| 37 | 8.68 | 8.53 | 0.15 | 8.86 | −0.18 |
| 38 | 8.41 | 8.58 | −0.17 | 8.56 | −0.15 |
| 39 | 8.96 | 8.93 | 0.03 | 8.96 | 0.00 |
| 41 | 8.92 | 8.91 | 0.01 | 9.00 | −0.08 |
| 42 | 9.49 | 9.34 | 0.15 | 9.25 | 0.24 |
| 43 | 5.54 | 5.83 | −0.29 | 5.80 | −0.26 |
| 44 | 5.85 | 5.72 | 0.13 | 5.88 | −0.03 |
| 45 | 6.43 | 5.92 | 0.51 | 5.92 | 0.51 |
| 46 | 5.33 | 5.58 | −0.25 | 5.58 | −0.25 |
| 47 | 5.82 | 6.03 | −0.21 | 6.29 | −0.47 |
| 48 | 5.59 | 6.13 | −0.54 | 6.01 | −0.42 |
| 49 | 6.52 | 6.20 | 0.32 | 6.12 | 0.40 |
| 50 | 6.82 | 6.70 | 0.12 | 6.28 | 0.54 |
| 51 | 6.13 | 6.29 | −0.16 | 6.34 | −0.21 |
| 52 | 6.37 | 6.37 | 0.00 | 6.26 | 0.11 |
| 54 | 5.68 | 5.76 | −0.08 | 5.67 | 0.01 |
| 55 | 5.89 | 5.77 | 0.13 | 6.20 | −0.31 |
| 56 | 8.36 | 8.17 | 0.19 | 8.18 | 0.18 |
| 57 | 7.92 | 7.99 | −0.07 | 7.87 | 0.05 |
| 59 | 8.34 | 8.22 | 0.12 | 8.13 | 0.21 |
| 60 | 8.12 | 8.10 | 0.02 | 8.10 | 0.02 |
| 61 | 6.72 | 6.79 | −0.07 | 6.51 | 0.21 |
| 62 | 6.77 | 6.64 | 0.13 | 6.84 | −0.07 |
| 63 | 8.17 | 8.12 | 0.05 | 8.16 | 0.01 |
| 65 | 8.29 | 8.06 | 0.23 | 8.31 | −0.02 |
| 66 | 7.82 | 8.01 | −0.19 | 8.09 | −0.27 |
| 67 | 8.08 | 8.13 | −0.05 | 8.28 | −0.20 |
| 69 | 8.82 | 8.48 | 0.34 | 8.60 | 0.22 |
| 70 | 8.57 | 8.47 | 0.10 | 8.47 | 0.11 |
| 72 | 8.48 | 8.59 | −0.11 | 8.43 | 0.05 |
| Compounds | Experimental pIC50 | CoMFA analysis | CoMSIA analysis | ||
|---|---|---|---|---|---|
| Predicted pIC50 | Residual | Predicted pIC50 | Residual | ||
| 1 | 6.80 | 6.93 | −0.13 | 6.45 | 0.35 |
| 2 | 6.96 | 5.21 | 1.75 | 5.89 | 1.07 |
| 12 | 7.38 | 7.56 | −0.18 | 7.19 | 0.19 |
| 21 | 6.42 | 6.70 | −0.28 | 6.94 | −0.52 |
| 24 | 8.70 | 8.38 | 0.32 | 8.26 | 0.44 |
| 28 | 6.54 | 6.95 | −0.41 | 6.88 | −0.34 |
| 35 | 7.60 | 7.58 | 0.02 | 7.75 | −0.15 |
| 40 | 9.17 | 9.18 | −0.01 | 9.08 | 0.09 |
| 53 | 5.64 | 6.01 | −0.37 | 6.13 | −0.49 |
| 58 | 8.19 | 8.31 | −0.12 | 8.70 | −0.51 |
| 64 | 7.70 | 8.04 | −0.34 | 8.50 | −0.80 |
| 68 | 8.09 | 8.20 | −0.11 | 8.48 | −0.39 |
| 71 | 8.85 | 8.51 | 0.34 | 8.88 | −0.03 |
| Compounds | Weighted pIC50 | CoMFA analysis | CoMSIA analysis | ||
|---|---|---|---|---|---|
| Predicted pIC50 | Residual | Predicted pIC50 | Residual | ||
| 6 | 7.67 | 7.40 | 0.28 | 7.05 | 0.62 |
| 11 | 7.90 | 8.15 | −0.25 | 7.95 | −0.05 |
| 12 | 8.58 | 8.91 | −0.33 | 8.60 | −0.02 |
| 13 | 8.70 | 8.93 | −0.23 | 8.66 | 0.04 |
| 15 | 8.12 | 8.35 | −0.23 | 8.62 | −0.50 |
| 16 | 9.79 | 10.31 | −0.52 | 9.74 | 0.05 |
| 17 | 8.64 | 8.74 | −0.10 | 8.98 | −0.34 |
| 19 | 8.50 | 8.57 | −0.07 | 8.83 | −0.33 |
| 20 | 9.44 | 9.85 | −0.41 | 9.71 | −0.27 |
| 21 | 6.98 | 7.28 | −0.30 | 6.98 | 0.00 |
| 22 | 9.41 | 9.56 | −0.15 | 9.57 | −0.16 |
| 23 | 9.61 | 10.12 | −0.51 | 10.06 | −0.45 |
| 24 | 9.98 | 10.23 | −0.25 | 10.32 | −0.33 |
| 26 | 10.53 | 11.01 | −0.48 | 11.09 | −0.56 |
| 28 | 7.33 | 7.45 | −0.12 | 7.90 | −0.57 |
| 29 | 8.00 | 8.07 | −0.07 | 8.00 | 0.00 |
| 31 | 8.14 | 7.92 | 0.22 | 7.98 | 0.16 |
| 33 | 7.14 | 7.46 | −0.32 | 7.32 | −0.18 |
| 37 | 9.48 | 9.68 | −0.20 | 9.68 | −0.20 |
| 38 | 9.34 | 9.64 | −0.30 | 9.34 | 0.00 |
| 39 | 10.33 | 10.51 | −0.18 | 10.60 | −0.27 |
| 40 | 10.64 | 11.00 | −0.36 | 11.06 | −0.41 |
| 42 | 10.50 | 10.44 | 0.06 | 10.57 | −0.06 |
| 45 | 5.57 | 5.48 | 0.09 | 6.00 | −0.43 |
| 49 | 7.20 | 6.95 | 0.25 | 6.88 | 0.33 |
| 50 | 7.67 | 7.86 | −0.19 | 7.41 | 0.26 |
| 51 | 6.61 | 6.69 | −0.08 | 6.74 | −0.13 |
| 52 | 7.18 | 7.48 | −0.30 | 6.89 | 0.29 |
| 53 | 6.21 | 6.50 | −0.29 | 6.55 | −0.34 |
| 56 | 9.52 | 9.91 | −0.39 | 9.67 | −0.15 |
| 57 | 8.85 | 9.25 | −0.40 | 9.30 | −0.45 |
| 60 | 9.33 | 9.64 | −0.31 | 9.88 | −0.55 |
| 61 | 7.77 | 8.07 | −0.30 | 8.44 | −0.67 |
| 62 | 7.99 | 8.90 | −0.91 | 8.14 | −0.15 |
| 63 | 9.18 | 9.27 | −0.09 | 9.17 | 0.01 |
| 64 | 8.75 | 9.10 | −0.35 | 8.74 | 0.01 |
| 67 | 9.32 | 9.49 | −0.17 | 9.55 | −0.23 |
| 68 | 9.47 | 9.65 | −0.18 | 9.99 | −0.51 |
| 70 | 9.62 | 9.55 | 0.07 | 9.68 | −0.06 |
| 71 | 9.98 | 9.85 | 0.14 | 10.18 | −0.20 |
| 72 | 9.12 | 9.45 | −0.33 | 9.36 | −0.24 |
| Compounds | Weighted pIC50 | CoMFA analysis | CoMSIA analysis | ||
|---|---|---|---|---|---|
| Predicted pIC50 | Residual | Predicted pIC50 | Residual | ||
| 14 | 8.37 | 8.63 | −0.26 | 8.14 | 0.23 |
| 18 | 9.36 | 10.24 | −0.88 | 9.88 | −0.52 |
| 25 | 10.01 | 9.71 | 0.30 | 9.73 | 0.28 |
| 27 | 9.29 | 10.04 | −0.75 | 10.01 | −0.72 |
| 32 | 8.43 | 8.04 | 0.39 | 8.11 | 0.32 |
| 34 | 8.32 | 8.20 | 0.12 | 8.47 | −0.15 |
| 35 | 8.40 | 7.87 | 0.53 | 7.92 | 0.48 |
| 36 | 9.10 | 8.73 | 0.37 | 8.58 | 0.52 |
| 41 | 10.14 | 9.85 | 0.29 | 10.37 | −0.23 |
| 58 | 9.03 | 9.34 | −0.31 | 9.35 | −0.32 |
| 59 | 9.44 | 9.53 | −0.09 | 9.26 | 0.18 |
| 65 | 9.44 | 9.81 | −0.37 | 9.20 | 0.24 |
| 66 | 9.01 | 9.32 | −0.31 | 9.55 | −0.54 |
| 69 | 10.17 | 9.65 | 0.52 | 9.81 | 0.36 |
| Model A | Model B | |||
|---|---|---|---|---|
| CoMFA | CoMSIA | CoMFA | CoMSIA | |
| No. compounds | 59 | 59 | 41 | 41 |
| Optimal number of components (ONC) | 6 | 6 | 6 | 6 |
| Leave one out r2 (rloo2) | 0.653 | 0.725 | 0.612 | 0.641 |
| Cross-validated r2 (rcv2) | 0.753 | 0.760 | 0.726 | 0.712 |
| Std. error of estimate (SEE) | 0.292 | 0.249 | 0.353 | 0.356 |
| Non cross-validated r2 (rncv2) | 0.946 | 0.961 | 0.968 | 0.968 |
| F value | 152.303 | 211.954 | 177.821 | 174.279 |
| Steric contribution | 0.592 | 0.216 | 0.529 | 0.193 |
| Electrostatic contribution | 0.408 | 0.193 | 0.471 | 0.206 |
| H bond acceptor contribution | — | 0.192 | — | 0.235 |
| H bond donor contribution | — | 0.110 | — | 0.106 |
| Hydrophobicity contribution | — | 0.289 | — | 0.260 |
| Bootstrap r2 (rboot2) | 0.962 | 0.987 | 0.981 | 0.975 |
| Standard error of estimate rboot2 (SEE rboot2) | 0.242 | 0.211 | 0.267 | 0.269 |
| Test set r2 (rpred2) | 0.73 | 0.77 | 0.71 | 0.77 |
The final model A CoMFA was generated by employing non-cross-validated PLS analysis with the optimum number of components (ONC = 6) to give a non-cross validated r2 (rncv2) = 0.946, a test set r2 (rpred2) = 0.73, Standard Error of Estimate (SEE) = 0.292, steric contribution = 0.592 and electrostatic contribution = 0.408.
The related CoMSIA analysis was derived using a statistical PLS analysis leading to the following results: ONC = 6, a non-cross validated r2 (rncv2) = 0.961, a test set r2 (rpred2) = 0.77, Standard Error of Estimate (SEE) = 0.249, steric contribution = 0.216, electrostatic contribution = 0.193, hydrophobic contribution = 0.289, H-bond acceptor = 0.192 and H-bond donor = 0.110.
An overall overview of the predictive ability of model A study can be obtained from graphical distributions of the predicted pIC50 values of the training set and test compounds, as shown in Fig. 4 and 5.
 | ||
| Fig. 4 Distribution of the predicted pIC50 values of the training set compounds with respect to the experimental data is shown, according to model A CoMFA and CoMSIA analyses. | ||
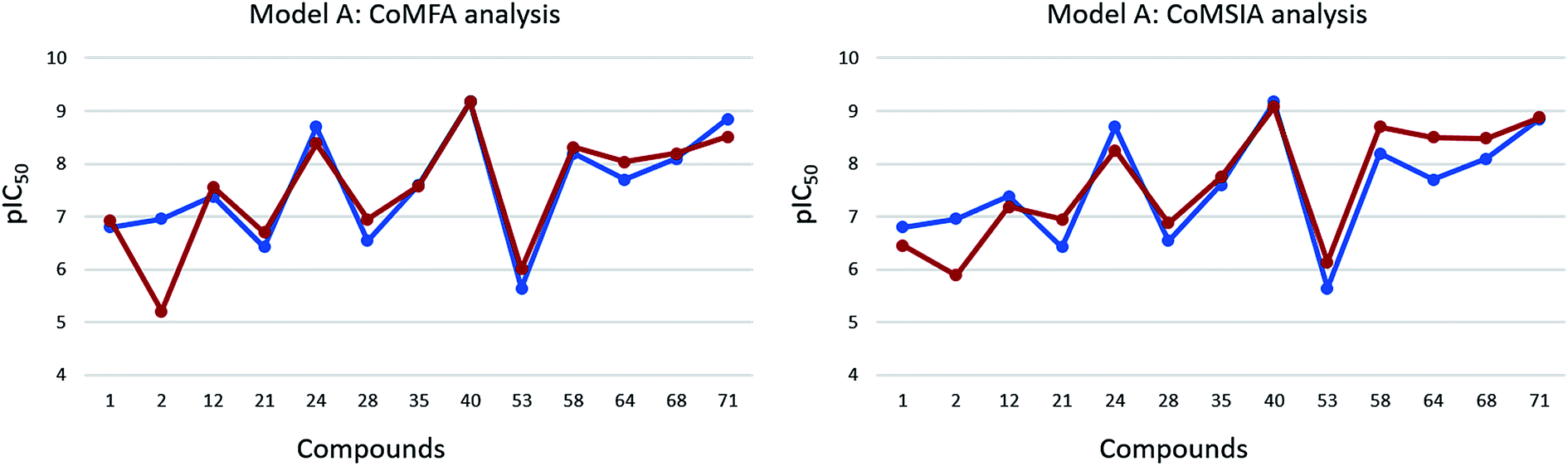 | ||
| Fig. 5 Representation of the experimental (blue dots and line) and predicted potency (dark orange dots and line) trend observed within the test set compounds, according to model A CoMFA and CoMSIA analyses. | ||
The selected CoMFA model B was generated by employing non-cross-validated PLS analysis with the optimum number of components (ONC = 6) to give a non-cross validated r2 (rncv2) = 0.968, a test set r2 (rpred2) = 0.71, Standard Error of Estimate (SEE) = 0.353, steric contribution = 0.529 and electrostatic contribution = 0.471. The CoMSIA model B was obtained with the following statistical results: ONC = 6, a non-cross validated r2 (rncv2) = 0.968, a test set r2 (rpred2) = 0.77, Standard Error of Estimate (SEE) = 0.356, steric contribution = 0.193, electrostatic contribution = 0.206, hydrophobic contribution = 0.260, H-bond acceptor = 0.235 and H-bond donor = 0.106.
The derived distributions of the predicted pIC50 values of the training set and test compounds are depicted in Fig. 6 and 7.
 | ||
| Fig. 6 Distribution of the predicted pIC50 values of the training set compounds with respect to the weighted one is shown, according to model B CoMFA and CoMSIA analyses. | ||
 | ||
| Fig. 7 Representation of the experimental-based (blue dots and line) and predicted weighted potency (dark orange dots and line) trend observed within the test set compounds, according to model B CoMFA and CoMSIA analyses. | ||
The CoMFA and CoMSIA model A and B reliability thus generated was also supported by bootstrapping results (see Table 6).
CoMFA steric and electrostatic maps
According to the CoMFA steric map descriptors, green polyhedra occupy those areas which prove to be favourable in terms of steric contacts while yellow maps highlight those regions related to unfavourable or slightly allowed steric interactions. On the basis of model A, as shown in Fig. 8a (compound 69 is shown as representative of the series of ligands), yellow polyhedra are placed in proximity of the R1 substituent, surround the R3 one while are placed quite far from the R2 group. On the other hand, a green area is located around the R2 group. According to a careful analysis of the whole dataset it can be observed that those compounds bearing a rigid group rather than a flexible one much more properly fit near the aforementioned yellow area in proximity of R1, explaining the higher potency trend of 28–32 (pIC50 = 5.64–7.39) with respect to 43–48 (pIC50 = 5.32–6.43). | ||
| Fig. 8 Contour maps of model A (a) and B (b) CoMFA steric regions (green, favoured; yellow, disfavoured) are shown around the PDE7 ligand 69 (C atom: white). The compound is displayed in ball and stick mode. | ||
Notably, this kind of information is quite in agreement with those obtained from the X-ray data and from docking calculations. Indeed, the R1 group should be projected towards the metal ion pocket, being included in a narrow region delimited by Thr321, Ile323 and Asp362 (see Fig. 2b).
On the contrary, most of the derivatives characterized by bulky groups at the R2 portion, efficiently arrange this substituent in the area delimited by the steric green polyhedra. Accordingly, a number compounds showing branched or hindered groups in R2 are endowed with a very high potency profile, as listed in Table 1 for compounds 22–27 (pIC50 = 7.92–9.00), 39–42 (pIC50 = 8.92–9.49) and 69–72 (pIC50 = 8.48–8.85). Conversely, the potency values of compounds 43–55 (pIC50 = 5.83–6.82), being unsubstituted at the R2 group, fall in lower pIC50 ranges if compared with the previously discussed analogues 22–27, 39–42 and 69–72.
Notably, the reliability of these results is supported by the following pIC50 trend: 37 (pIC50 = 8.68) > 36 (pIC50 = 8.08) > 35 (pIC50 = 7.60) > 34 (pIC50 = 7.22).
In addition, these results are supported by the docking poses previously discussed, being the R2 substituent oriented toward a deeper pocket, including F384, L401 and L420, than that occupied by R1.
The CoMFA model developed on the basis of the selectivity issue (model B) better underlines a key role played by bulky groups at the R2 substituent (see Fig. 8b), and also allows the introduction of much more flexible portions in the area occupied by R1 (smaller yellow polyhedral in comparison to model A). Interestingly, all these results are supported by the PDE7 and PDE4B potency trend observed moving from compounds 11 (PDE7 pIC50 = 7.01; PDE4B pIC50 = 5.42), 24 (PDE7 pIC50 = 8.70; PDE4B pIC50 = 6.42), 61 (PDE7 pIC50 = 6.72; PDE4B pIC50 = 4.85) to the related (much more bulky in R2) 12 (PDE7 pIC50 = 7.38; PDE4B pIC50 = 5.23), 26 (PDE7 pIC50 = 9.00; PDE4B pIC50 = 6.28), 62 (PDE7 pIC50 = 6.77; PDE4B pIC50 = 4.60).
As a consequence, this steric map emphasizes the importance of proper hindered portions, such as bicyclic and/or aromatic moieties, at these positions achieving selectivity to PDE7 over PDE4B.
Accordingly, as described about the compounds docking mode within the PDE4B isoform, any substituent placed in R1 falls at a narrow region, while the one in R2 resulted to be quite far from the metal binding pocket.
The CoMFA electrostatic descriptors are shown as blue areas around those regions predicted to be beneficial for electropositive moieties, while red polyhedra occupy any area recommended for much more electronegative groups.
For model A, the electrostatic CoMFA map reported in Fig. 9a revealed a large blue area, surrounding a consistent red one, in proximity of the R1 substituent.
 | ||
| Fig. 9 Contour maps of model A (a) and B (b) CoMFA electrostatic regions are shown around the PDE7 ligand 69 (C atom: white). The compound is displayed in ball and stick mode. Blue regions are favourable for more positively charged groups; red regions are favourable for less positively charged groups. | ||
The reliability of these results is supported by the higher potency values of those compounds bearing electron-rich portions in R1, such as aromatic amines, rather than the aliphatic ones [compare 28–34 (pIC50 = 5.64–7.39) with 43–55 (pIC50 = 5.33–6.52)].
A second small blue map is also located in proximity of a methylene of the R2 pyrrolidine ring of compound 69, while the amide portion of the same substituent properly falls in a red area. In addition, any group eventually connected to the first part of the R2 substituent (that is directly linked to the thieno[3,2-d]pyrimidin-4(3H)-one core) is surrounded by favourable areas for electropositive moieties (blue polyhedral).
These data suggest a favourable role played by the introduction of an electron-rich nucleus in R2, eventually decorated with proper polar substituents. This information is in agreement with the high potency profile displayed by those derivatives bearing aromatic features and/or aliphatic groups connected with amide or polar moieties [see 22–26 (pIC50 = 8.30–9.00) and 37–42 (pIC50 = 8.41–9.49)], and also by the lower potency values of those compounds being unsubstituted in R2 [see 8–10 (pIC50 = 5.19–5.96).
The electrostatic profile here discussed about R1 and R2 is strongly in agreement with the information coming from the experimental X-ray data and docking studies, that reveal the role of these substituents in water-bridges contacts, being also (the most bulky of them) oriented toward the catalytic site metal binding pocket.
Concerning the selectivity issue (Fig. 9b), the electrostatic map disclosed by model B underlines the relevance of proper electrostatic features around R1. In particular, the presence of an electron-rich substituent, surrounded by electropositive features results to be beneficial to achieve higher selectivity profile toward PDE7 over PDE4B. On these basis, the introduction of an aliphatic or cyclic aliphatic amine in R1 results to be much more promising than that of an aromatic one. It should be noticed that this information is supported by the results obtained from docking studies around PDE4B. Indeed, based on the observed docking mode of 6 in Fig. 3, an aliphatic R1 group could be in clash with the narrow pocket delimited by Met431 and Ile450, leading to much more selective PDE7 inhibitors.
Accordingly, a number of cyclopentanamine-based analogues [such as 16–20 (PDE7 pIC50 = 7.41–8.38; PDE4B pIC50 = 5.47–6.17) and 23–27 (PDE7 pIC50 = 7.92–9.00; PDE4B pIC50 = 5.48–6.42)], and also of propan-2-amine based compounds [such as 60–68 (PDE7 pIC50 = 6.72–8.29; PDE4B pIC50 = 4.60–6.36)], are endowed with a marked selectivity profile to PDE7 over PDE4B.
CoMSIA hydrophobic, H-bond acceptor and H-bond donor maps
The CoMSIA hydrophobic map reveals any ligand feature predicted to be favoured (yellow polyhedra) for lipophilic groups, while white areas represent the ligand portion to be decorated with more polar moieties.Taking into account the CoMSIA hydrophobic map derived from model A (see Fig. 10a), more lipophilic groups are predicted to be beneficial at the first portion of the R1 substituent and in the area placed in proximity of the central core of R2. Notably, the reliability of this information is supported by the following potency trend: 32 (R1 = 2-chloroaniline; pIC50 = 7.39) > 31 (R1 = 2-fluoroaniline; pIC50 = 7.21) > 29 (R1 = 2-methylaniline; pIC50 = 7.06) > 28 (R1 = aniline; pIC50 = 6.54) > 30 (R1 = 2-aminobenzonitrile; pIC50 = 5.64). In addition, these data are also supported by the lower potency displayed by compounds 53–55 (pIC50 = 5.64–5.89) if compared with the analogue 51 (pIC50 = 6.13).
 | ||
| Fig. 10 CoMSIA hydrophobic favoured (yellow area) and disfavoured (grey area) regions calculated through model A (a) and model B (b) are shown around compound 69 (C atom: white). The compound is displayed in ball and stick mode. | ||
On the other hand, the introduction of less hydrophobic groups is recommended in proximity of the terminal region of R1 and R2 substituent (near the nitrogen atom of the pyridine ring of compound 69, as shown in Fig. 10a), being in accordance with the ability to interact with water molecules and to move near the enzyme metal ions, as previously discussed.
In addition, this data are also in agreement with the high pIC50 values of compounds 39–42 (pIC50 = 8.92–9.49) bearing in R2 a slightly lipophilic linker connecting polar functions with the thieno[3,2-d]pyrimidin-4(3H)-one core.
Finally, the presence of hydrophobic groups is discouraged around R3 (white polyhedral). Accordingly, compound 4 (pIC50 = 5.17) is characterized by lower potency value than compounds 1–3 (pIC50 = 6.51–6.96).
According to the CoMSIA hydrophobic map described by model B (see Fig. 10b), the presence of lipophilic groups results to be particularly beneficial at the final part of the R1 substituent, rather than at its first portion, and at the central area occupied by the R2 group.
In agreement with these data, it should be noticed an overall much more effectiveness in terms of selectivity, moving from compounds 49–51 (PDE7 pIC50 = 6.13–6.82; PDE4B pIC50 = 5.28–5.32), bearing a cycloaliphatic amine in R1, to the analogue 53 (PDE7 pIC50 = 5.64; PDE4B pIC50 = 4.62), showing a 4-(methylamino)cyclohexanol substituent in R1.
The reliability of this kind of analysis is also in accordance with the promising selectivity trend observed for compounds 56–60 (PDE7 pIC50 = 7.92–8.36; PDE4B pIC50 = 5.96–6.70), bearing an aromatic feature in R2.
Concerning the H-bond acceptor map obtained from model A (Fig. 11a), unfavourable H bond acceptor contours (green areas) are reported in proximity of the R1 substituent, in particular near the nitrogen atom of compound 69, therefore discouraging the introduction at this position of any feature able to behave as H-bond acceptor.
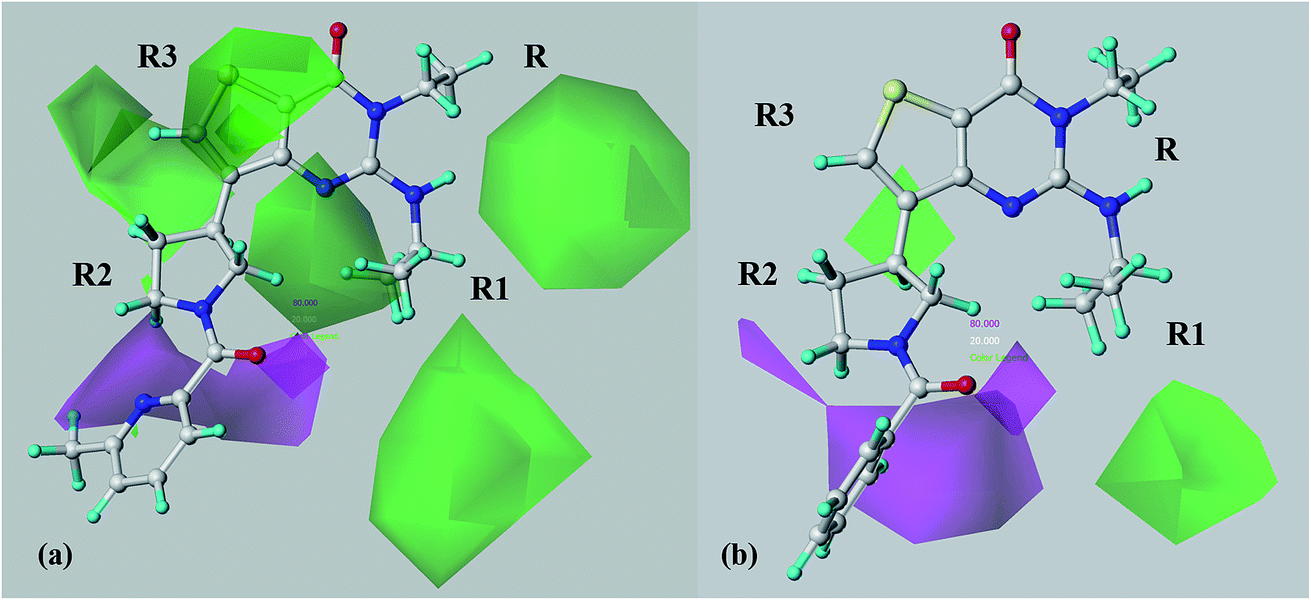 | ||
| Fig. 11 CoMSIA H-bond acceptor favoured (magenta area) and disfavoured (red area) contour maps derived from model A (a) and model B (b) are shown around compound 69 (C atom: white). The compound is displayed in ball and stick mode. | ||
Interestingly, this kind of information probably derives from the privileged role (in terms of potency level to PDE7) played by the aromatic amines (which can only act as H-bond donor moieties) rather than the aliphatic ones (which can act both as H-bond donor and acceptor groups), as R1 substituent. The reliability of these results is supported by the higher pIC50 values of the aniline and 2-fluoroaniline derivatives 28 (pIC50 = 6.54) and 31 (pIC50 = 7.21) if compared with most of the aliphatic amine analogues, such as 43–48 (pIC50 = 5.33–6.43).
In addition, other green polyhedra are placed in proximity of the terminal part of R1, near the portion of the R2 substituent that is directly linked to the thieno[3,2-d]pyrimidin-4(3H)-one core, and also around the R3 substituent. Concerning R1, this data are in agreement with the poor potency profile of compounds 52–55 (pIC50 = 5.64–6.37), showing additional H-bond acceptor moieties at this position.
A consistent region of the central portion of R2 is surrounded by favourable areas for H-bond acceptor functions (depicted in magenta), suggesting a positive relevance played by at least one (or also two) H-bond acceptor pharmacophore feature(s) in R2. Accordingly, in the case of molecules showing two H-bond acceptor groups, compounds 26 (pIC50 = 9.00), 41 (pIC50 = 8.92) and 42 (pIC50 = 9.49) display very promising PDE7 potency levels.
It should be noticed that these data are verified by the docking results previously mentioned, supporting the role of bulky R2 substituents H-bonded with the Ile323 backbone.
Finally, these results are also in accordance with the high pIC50 values of those compounds bearing one H-bond acceptor moiety in R2, such as an amide function linked through a proper spacer to the thieno[3,2-d]pyrimidin-4(3H)-one core [see 67–72 (pIC50 = 8.08–8.85)].
According to the H-bond acceptor map derived from model B (Fig. 11b), the selectivity profile of this series of molecules proves to be related with the presence of proper substituents in R2 (bearing one or also two H-bond acceptor moieties) which could fully project toward a consistent magenta polyhedral. Indeed, the H-bonding R2 groups of thieno[3,2-d]pyrimidin-4(3H)-one derivatives (as PDE7 inhibitors) are oriented in proximity of the catalytic site metal binding pocket, while at the PDE4B enzyme are arranged in a different way, being much more far from the mentioned metal ions.
This kind of map can be easily and efficiently occupied both by rigid and by flexible decorations. This information is supported by the high selectivity ratio observed for compounds 25 (PDE7 pIC50 = 8.64; PDE4B pIC50 = 6.19) if compared with that of the related 26 (PDE7 pIC50 = 9.00; PDE4B pIC50 = 6.28).
Notably, the introduction of H-bond acceptor groups at the first part of the R1 substituent results to be allowed, therefore revealing an overall positive effect played both by the aliphatic amines and by the aromatic ones at this position. Conversely, the presence of additional H-bond acceptor groups results to be detrimental for selectivity, as previously observed for the model A H-bond acceptor map.
Taking into account an overall analysis of the H-bond donor features highlighted by the model A and model B calculations (Fig. 12), the PDE7 potency trend of this series of inhibitors increases in absence of H-bond donor group at the R2 substituent (purple area), being in agreement with the related information coming from the H-bond acceptor map. On the other hand, in particular the selectivity profile seems to be influenced by the availability of H-donor group in R1 (cyan area), being in any case in accordance with the results we previously discussed about this kind of substituent (model B H-bond acceptor map).
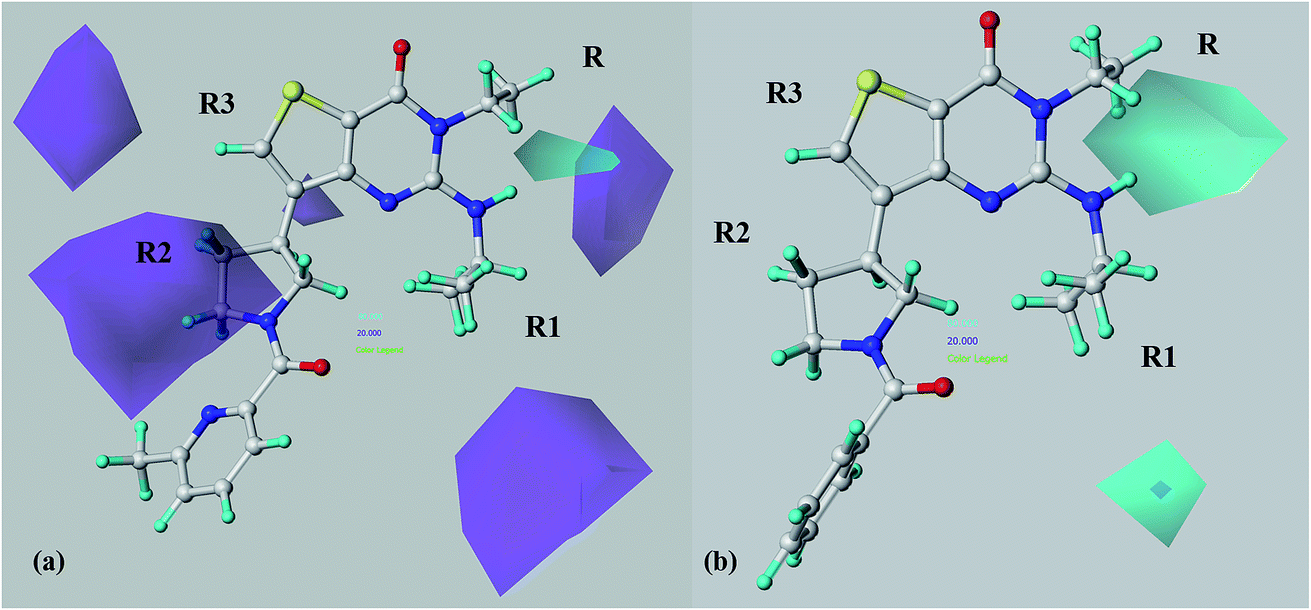 | ||
| Fig. 12 CoMSIA H-bond donor favoured (cyan area) and disfavoured (purple area) contour maps calculated by model A (a) and model B (b) are depicted around compound 69 (C atom: white). The compound is displayed in ball and stick mode. | ||
With the aim at gaining a qualitative validation of the results here discussed, we also performed a comparison with external series of PDE7 inhibitors, designed around different chemical scaffold, which proved to be isostere or bio-isostere of the thieno-pyrimidinone one. In particular, we evaluated the ability of the derived 3D-QSAR maps to rationalize the potency and selectivity profile of dihydronaphthyridinediones, and isothiazole- or isoxazole-fused pyrimidines.32,33
Thus, the topology and the molecular electrostatic potential displayed on the Connolly surface of compound I revealed a comparable distribution of the positive- and negative-charged regions, depicted in blue and red, onto the dihydronaphthyridinedione ring with respect to the thieno-pyrimidinone one (Fig. 13). On this basis, the I difluorophenyl ring mimics the compound 26 R1 group, unfortunately lacking of any H-bonding moiety to be exploited for water-bridges contacts. In addition, this compound is unsubstituted at the bicyclic position 8, that is the one that could probably simulate the role played by the R2 group of the inhibitor 26. Accordingly, it was endowed with a lower potency ability than 26. Nevertheless, this dihydronaphthyridinedione derivative displays an acceptable selectivity trend within PDE7, being able to fulfil the model B CoMSIA hydrophobic map (suggesting a flexible and hydrophobic substituent in R1).
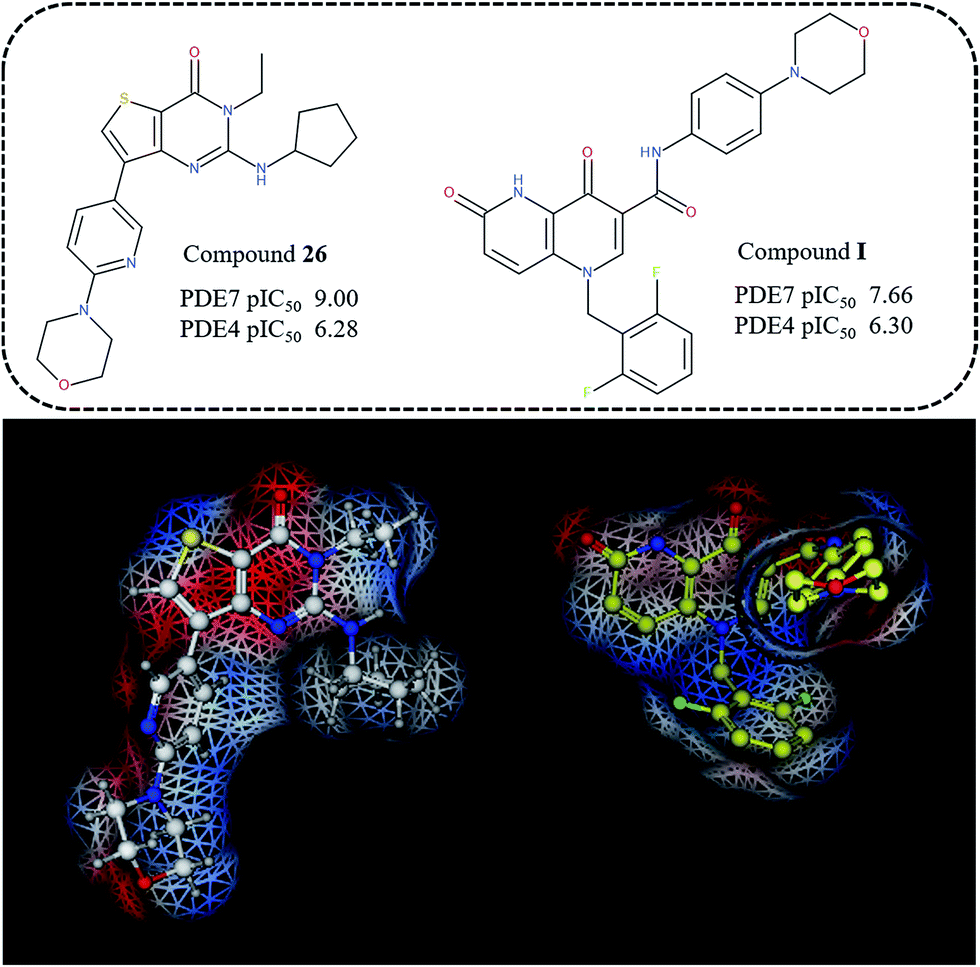 | ||
| Fig. 13 Representation of the molecular electrostatic potential around compounds 6 and I. | ||
The results derived around the isothiazole- or isoxazole-fused pyrimidines are quite comparable and, as shown in Fig. 14, reveal a good accordance between the molecular electrostatic profile evaluated along the substituent place at the compound II isothiazole-fused pyrimidine position 2 and the one in R2 (for compound 26), lacking of any group R1-like to properly satisfy any requirement described by model 3D-QSAR studies.
 | ||
| Fig. 14 Representation of the molecular electrostatic potential around compounds 6 and II. | ||
Conclusions
The computational studies here presented highlight and discuss the role played by the steric and electrostatic features, and also those due to hydrophobic, H-bond acceptor and donor moieties, in terms of PDE7 inhibitor pharmacophore. Through 3D-QSAR studies, any pattern of requirements specifically related to optimized PDE7 potency and selectivity trends was addressed and deeply discussed, even in the case to be applied around other bioisostere rings. The information coming from CoMFA and CoMSIA studies could pave the way for a more focused development of new compounds, endowed with improved potency and selectivity toward PDE7.Experimental
Ligand preparation
For our calculation, a pool of 72 selective PDE7A inhibitors was chosen from those available in literature. All compounds were built in silico and energy minimized within MOE using MMFF94 force field.34 Therefore, all the compounds were also parameterized by means of the Gasteiger–Hückel method.Calculation of molecular electrostatic potential was performed based on the AM1 charge system within MOE software.
Docking studies
All the compounds have been docked into the enzyme catalytic site of the PDE7 X-ray crystallographic data 4Y2B and PDE4B (pdb code 3GWT), by means of the Surflex docking module implemented in Sybyl-X1.0.35 Then, for all the compounds, the best docking geometries of both the ligand and the closer amino acid residues (selected by means of the SurFlex scoring functions) were refined by ligand/protein complex energy minimization implemented in Sybyl-X1.0. All calculations were carried out using a PC running the Windows XP operating system.3D-QSAR analyses
Model A and model B CoMFA and CoMSIA analyses were performed to better understand especially how the steric and electrostatic profiles so ad hydrophobic, H-bond acceptor and H-bond donor features, influence the potency and selectivity trend of this series of compounds with respect to the PDE7 enzyme.Training set and test set
All the compounds were grouped into a training set, for model generation, and a test set, for model validation, containing: (i) 59 and 13 compounds for model A; (ii) 41 and 14 inhibitors for model B; respectively. In any model, the molecules selected for the training and the test set pools were chosen manually, based on representative criteria of the overall biological activity trend and structural variations.For model A and model B analyses, IC50 values have been transformed into pIC50 values and then used as response variables.
In particular, for model B the inhibitor potency values were re-calculated by taking into account the difference in pIC50 between the PDE7 and PDE4B isoforms, obtaining a sort of weighted PDE7 pIC50, according to the following equation:
| Weighted PDE7 pIC50 = PDE7 pIC50 + [(PDE7 pIC50 − PDE4B pIC50)/MD] |
Notably, it is expected that such a weighted parameter could be much more predictive of the inhibitor selectivity profile observed within the chemical space disclosed around the thieno[3,2-d]pyrimidin-4(3H)-one derivatives here investigated.
CoMFA and CoMSIA models and statistical evaluation
CoMFA and CoMSIA methods are widely used 3D-QSAR techniques being useful to relate any variation of the experimental biological activity of a series of compounds (dependent variables) with respect to specific descriptors (independent variables). In details, the steric and electrostatic fields and especially the hydrophobic, H-bond donor, H-bond acceptor ones were employed for CoMFA and CoMSIA analysis, respectively. Starting from a proper molecule alignment within a 3D cubic lattice (with a 2 Å grid spacing), the any descriptor was calculated, using the standard Tripos force field method. Successively, the derived model goodness and reliability were evaluated using specific statistical tools, such as partial least square (PLS) analysis and cross-validation methods.Finally, the predictive ability concerning those compounds belonging to the test set (rpred2) was also calculated, by means of the following equation:
| rpred2 = (SD − PRESS)/SD |
Any further detail concerning the (standard) CoMFA and CoMSIA procedures and the statistical and predictive evaluation we applied, were previously reported in a consistent number of our works.36–41
Conflict of interest
The authors declare that they have no conflict of interest.Acknowledgements
This work was financially supported by the University of Genoa. Authors would like to thank Mr V. Ruocco for the informatic support to calculations.References
- A. T. Bender and J. A. Beavo, Pharmacol. Rev., 2006, 58, 488–520 CrossRef CAS PubMed.
- J. A. Beavo and R. J. Heaslip, Mol. Pharmacol., 1994, 46, 399–405 CAS , ASPET meeting report.
- F. Numano, Y. Kishi and T. Ashikaga, Adv. Second Messenger Phosphoprotein Res., 1992, 25, 395–407 CAS.
- M. J. Perry and G. A. Higgs, Curr. Opin. Chem. Biol., 1998, 4, 472–481 CrossRef.
- J. Polson and S. J. Strada, Annu. Rev. Pharmacol. Toxicol., 1996, 36, 403–427 CrossRef CAS PubMed.
- D. P. Rotella, Nat. Rev. Drug Discovery, 2002, 9, 674–682 CrossRef PubMed.
- S. Grundy, J. Plumb, M. Kaur, D. Ray and D. Singh, Respir. Res., 2016, 17, 9, DOI:10.1186/s12931-016-0325-8.
- N. J. Press, R. J. Taylor, J. D. Fullerton, P. Tranter, C. McCarthy, T. H. Keller, N. Arnold, D. Beer, L. Brown, R. Cheung, J. Christie, A. Denholm, S. Haberthuer, J. D. Hatto, M. Keenan, M. K. Mercer, H. Oakman, H. Sahri, A. R. Tuffnell, M. Tweed and A. Trifilieff, J. Med. Chem., 2015, 17, 6747–6752 CrossRef PubMed.
- A. B. Burgin, O. T. Magnusson, J. Singh, P. Witte, B. L. Staker, J. M. Bjornsson, M. Thorsteinsdottir, S. Hrafnsdottir, T. Hagen, A. S. Kiselyov, L. J. Stewart and M. E. Gurney, Nat. Biotechnol., 2010, 1, 63–70 CrossRef PubMed.
- M. Torras-Llort and F. Azorín, Biochem. J., 2003, 373(3), 835–843 CrossRef CAS PubMed.
- R. Lee, S. Wolda, E. Moon, J. Esselstyn, C. Hertel and A. Lerner, Cell. Signalling, 2002, 3, 277–284 CrossRef.
- P. Wang, P. Wu, R. W. Egan and M. M. Billah, Biochem. Biophys. Res. Commun., 2000, 3, 1271–1277 CrossRef PubMed.
- J. M. Hetman, S. H. Soderling, N. A. Glavas and J. A. Beavo, Proc. Natl. Acad. Sci. U. S. A., 2000, 1, 472–476 CrossRef.
- S. J. Smith, S. Brookes-Fazakerley, L. E. Donnelly, P. J. Barnes, M. S. Barnette and M. A. Giembycz, Am. J. Physiol.: Lung Cell. Mol. Physiol., 2003, 2, L279–L289 CrossRef PubMed.
- M. Safavi, M. Baeeri and M. Abdollahi, Expert Opin. Drug Discovery, 2013, 6, 733–751 CrossRef PubMed.
- H. Dong, C. Zitt, C. Auriga, A. Hatzelmann and P. M. Epstein, Biochem. Pharmacol., 2010, 79, 321–329 CrossRef CAS PubMed.
- M. Pekkinen, M. E. Ahlström, U. Riehle, M. M. Huttunen and C. J. Lamberg-Allardt, Bone, 2008, 43, 84–91 CrossRef CAS PubMed.
- S. Yamamoto, S. Sugahara, K. Ikeda and Y. Shimizu, Eur. J. Pharmacol., 2006, 550, 166–172 CrossRef CAS PubMed.
- S. Yamamoto, S. Sugahara, R. Naito, A. Ichikawa, K. Ikeda, T. Yamada and Y. Shimizu, Eur. J. Pharmacol., 2006, 541, 106–114 CrossRef CAS PubMed.
- K. Kawai, Y. Endo, T. Asano, S. Amano, K. Sawada, N. Ueo, N. Takahashi, Y. Sonoda, M. Nagai, N. Kamei and N. Nagata, J. Med. Chem., 2014, 23, 9844–9854 CrossRef PubMed.
- Y. Endo, K. Kawai, T. Asano, S. Amano, Y. Asanuma, K. Sawada, K. Ogura, N. Nagata, N. Ueo, N. Takahashi, Y. Sonoda and N. Kamei, Bioorg. Med. Chem. Lett., 2015, 3, 649–653 CrossRef PubMed.
- Y. Endo, K. Kawai, T. Asano, S. Amano, Y. Asanuma, K. Sawada, Y. Onodera, N. Ueo, N. Takahashi, Y. Sonoda, N. Kamei and T. Irie, Bioorg. Med. Chem. Lett., 2015, 9, 1910–1914 CrossRef PubMed.
- S. Guariento, O. Bruno, P. Fossa and E. Cichero, Mol. Diversity, 2016, 1, 77–92 CrossRef PubMed.
- Z. Ul-Haq, N. Khan, S. K. Zafar and S. T. Moin, Eur. J. Pharm. Sci., 2016, 88, 26–36 CrossRef CAS PubMed.
- P. K. Balasubramanian, A. Balupuri and S. J. Cho, Arch. Pharm. Sci. Res., 2016, 39, 328–339 CrossRef CAS PubMed.
- S. A. Halim and Z. Ul-Haq, Chem.-Biol. Interact., 2015, 238, 9–24 CrossRef CAS PubMed.
- E. Cichero, O. Bruno and P. Fossa, J. Enzyme Inhib. Med. Chem., 2012, 5, 730–743 CrossRef PubMed.
- E. Cichero, S. Cesarini, P. Fossa, A. Spallarossa and L. Mosti, Eur. J. Med. Chem., 2009, 5, 2059–2070 CrossRef PubMed.
- M. D. Woodrow, S. P. Ballantine, M. D. Barker, B. J. Clarke, J. Dawson, T. W. Dean, C. J. Delves, B. Evans, S. L. Gough, S. B. Guntrip, S. Holman, D. S. Holmes, M. Kranz, M. K. Lindvaal, F. S. Lucas, M. Neu, L. E. Ranshaw, Y. E. Solanke, D. O. Somers, P. Ward and J. O. Wiseman, Bioorg. Med. Chem. Lett., 2009, 17, 5261–5265 CrossRef PubMed.
- R. D. Cramer III, D. E. Patterson and J. D. Bunce, J. Am. Chem. Soc., 1988, 110, 5959–5967 CrossRef PubMed.
- G. Klebe, U. Abraham and T. Mietzner, J. Med. Chem., 1994, 37, 4130–4146 CrossRef CAS PubMed.
- R. Gewald, C. Rueger, C. Grunwald, U. Egerland and N. Hoefgen, Bioorg. Med. Chem. Lett., 2011, 21, 6652–6656 CrossRef CAS PubMed.
- A. Banerjee, P. S. Yadav, M. Bajpai, R. R. Sangana, S. Gullapalli, G. S. Gudi and L. A. Gharat, Bioorg. Med. Chem. Lett., 2012, 22, 3223–3228 CrossRef CAS PubMed.
- MOE, Chemical Computing Group Inc., Montreal, H3A 2R7, Canada, http://www.chemcomp.com.
- Sybyl-X 1.0, Tripos Inc., 1699 South Hanley Road., St Louis, Missouri, 63144, USA Search PubMed.
- E. Cichero, A. Ligresti, M. Allarà, V. Di Marzo, Z. Lazzati, P. D'Ursi, A. Marabotti, L. Milanesi, A. Spallarossa, A. Ranise and P. Fossa, Eur. J. Med. Chem., 2011, 46, 4489–4505 CrossRef CAS PubMed.
- E. Cichero, L. Buffa and P. Fossa, J. Mol. Model., 2011, 7, 1537–1550 CrossRef PubMed.
- E. Cichero, S. Cesarini, L. Mosti and P. Fossa, J. Mol. Model., 2010, 16, 1481–1498 CrossRef CAS PubMed.
- E. Cichero, S. Cesarini, L. Mosti and P. Fossa, J. Mol. Model., 2010, 16, 677–691 CrossRef CAS PubMed.
- E. Cichero, S. Cesarini, P. Fossa, A. Spallarossa and L. Mosti, J. Mol. Model., 2009, 15, 871–884 CrossRef CAS PubMed.
- E. Cichero and P. Fossa, J. Mol. Model., 2012, 18, 1573–1582 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra12624c |
| This journal is © The Royal Society of Chemistry 2016 |