
Protein binding-protected DNA three-way junction-mediated rolling circle amplification for sensitive and specific detection of transcription factors†
Kan Lia,
Lei Wangb,
Xiaowen Xua,
Ting Gaoa,
Ping Yan*c and
Wei Jiang*a
aKey Laboratory for Colloid and Interface Chemistry of Education Ministry, School of Chemistry and Chemical Engineering, Shandong University, 250100 Jinan, P. R. China. E-mail: wjiang@sdu.edu.cn; Fax: +86 531 88564464; Tel: +86 531 88363888
bSchool of Pharmaceutical Sciences, Shandong University, 250012 Jinan, P. R. China
cJinan Maternity and Child Care Hospital, 250001 Jinan, P. R. China. E-mail: yanping6768@163.com
First published on 6th July 2016
Abstract
Transcription factors (TFs) are DNA-binding proteins that regulate gene transcription and their expression levels are closely associated with disease development. Sensitive and specific detection of TFs is significant to clinical diagnostics and drug development. Herein, a label-free fluorescent strategy for the sensitive and specific detection of TFs was developed based on protein binding-protected DNA three-way junction (TWJ)-mediated rolling circle amplification (RCA). A trifunctional TWJ was designed including a target binding site, AlwI recognition site and sutured primer of RCA. Firstly, TFs bound with a target binding site, protecting the four and five bases downstream from the AlwI recognition site against cleavage by AlwI. Next, the sutured primer in the protected TWJ hybridized with the padlock probe, initiating RCA. Finally, the sutured primer was extended with multiple G-quadruplex sequences, binding with N-methyl-mesoporphyrin IX (NMM) to yield an enhanced fluorescence response. Residual TWJs were digested by AlwI, effectively blocking the RCA reaction, thus suppressing nonspecific amplification. Taking NF-κB p50 as a model target of TFs, high sensitivity was achieved with a low detection limit of 6.8 pM and a broad linear range from 8 pM to 15 nM. We successfully measured NF-κB p50 in HeLa cell nuclear extracts with a low detection limit of 0.34 ng μL−1. The strategy was also effectively used to assay the inhibition effect of a model inhibitor of NF-κB, oridonin. The results indicated the proposed strategy holds great promise for studying TFs in disease diagnostics and drug developments.
1. Introduction
Transcription factors (TFs) are important DNA-binding proteins that modulate genetic transcription via binding with specific DNA sequences within the gene regulatory region.1,2 Regular expression of TFs is essential for normal cellular growth and survival,3,4 whereas dysregulation might lead to multiple diseases such as autoimmune diseases,5 congenital heart disease,6 diabetes,7 and cancer.8 Sensitive and specific detection of TFs is significant to study the critical function of TFs in gene expression and early diagnosis of various diseases.The conventional methods for TFs detection are DNA footprinting assay,9 western blotting assay,10 electrophoretic mobility shift assay (EMSA)11 and enzyme-linked immunosorbent assay (ELISA).12 However, the radioactive hazards and the instability of antibody limit their extensive applications. Various fluorescence-based assays have been developed due to their safety and simplicity. Currently, two categories of fluorescence-based assays for TFs detection have been developed. One category is based on the conformational transition of recognition probe.13–15 For example, Heyduk et al. have developed a bimolecular proximity assay based on two duplexes containing half of the TFs binding sequence, which generated combination with the target protein binding.13 Plaxco and his co-workers have reported a method for TFs detection employing a double stem-loop structure which generated conformational equilibrium shift to single stem-loop structure with the specific binding of TFs.15 Nevertheless, the split of binding site affected the binding affinity of target protein, reducing the detection efficiency. And the conformational equilibrium of recognition probe was susceptible to the environmental factors, leading to false positive results. The other category is based on nuclease cleavage-inhibition assay,16–20 avoiding the split of binding site or conformational equilibrium of recognition probe. In the presence of TFs, the DNA probe can be protected from digestion by nuclease due to the steric hindrance of protein binding. For example, Wang's group have developed a method using exonuclease III (Exo III) to distinguish TFs-binding DNA probe from others.16 However, Exo III can replace some DNA-binding TFs, leading to undesired cleavage of DNA probes, which has been verified by Yu and his co-workers.21 Thus false negative results may be obtained, reducing the detection sensitivity. In addition, Wang et al. have taken advantage of Apo I, a cleavage sequence-specific enzyme, to achieve TFs detection.20 The DNA probe labelled with fluorescence donor and receptor can be protected efficiently once the particular cleavage sequence is occupied by TFs, with high FRET. Unfortunately, it required the binding site of TFs with a specific sequence for enzyme cleavage, which limited its versatility. The labelling of the DNA probe also reduced the binding affinity of TFs. To address the above issues, the development of a label-free fluorescent strategy for sensitive, specific and versatile detection of TFs is still highly desirable.
Herein, we developed a label-free fluorescent strategy based on protein binding-protected DNA three-way junction (TWJ)-mediated rolling circle amplification (RCA) reaction for sensitive, specific and versatile detection of TFs. Firstly, TFs bound with the target protein binding site, protecting the four and five bases downstream from AlwI recognition site against cleavage by AlwI, an unusual enzyme that recognizes a specific DNA sequence 5′-GGATC-3′ and precisely cleaves a short distance outside the recognition site.22,23 Next, the sutured primer in the protected TWJ hybridized with the padlock probe. Both ends of padlock probe were specifically ligated in the presence of T4 ligase and the circular template was formed. Then, with the addition of phi29 DNA polymerase, the RCA reaction was initiated. RCA is an isothermal DNA replication technique which can generate a large number of tandem DNA repeats complementary to the circular template in hours.24 In this case, the sutured primer was extended with a long G-quadruplex (G4) sequence. Finally, the G4 sequence bound with N-methyl-mesoporphyrin IX (NMM) to yield a label-free and enhanced fluorescence response.25,26 Residual TWJs were digested by AlwI with the sutured primer broken, effectively blocking RCA reaction, thus suppressing nonspecific amplification. Taking NF-κB p50 as a model target of TFs, high sensitivity was achieved with a low detection limit of 6.8 pM and a broad linear range from 8 pM to 15 nM. We successfully measured the target TF in HeLa cell nuclear extracts with a low detection limit of 0.34 ng μL−1. The strategy was also effectively used to assay the inhibition effect of a model inhibitor of NF-κB, oridonin. The results indicated the proposed strategy held great promise for studying of TFs in disease diagnostics and drug developments.
2. Experimental section
2.1. Reagents and materials
DNA oligonucleotides were synthesized and purified by Sangon Inc. (Shanghai, China). The DNA sequences were listed in Table S1.† NF-κB p50 was obtained from Cayman Chemical (Ann Arbor, MI, USA). AlwI was provided by New England Biolabs, Ltd. (Beverly, MA, USA). T4 DNA ligase, phi29 DNA polymerase, bovine serum albumin (BSA) and deoxynucleotides (dNTPs) were obtained from Sangon Inc. (Shanghai, China). NMM and SYBR Green I was provided by Frontier Scientific Inc. (Utah, USA) and BioTeke Corporation (Beijing, China), respectively. Carcinoembryonic antigen (CEA), c-Jun protein and folate receptor (FR) were provided by Sino Biological Inc. (Beijing, China), Linc-Bio Science Co. Ltd. (Shanghai, China) and Cayman Chemical (Ann Arbor, MI), respectively. We purchased TNF-α-treated HeLa nuclear extracts from Active Motif (Carlsbad, USA). All other reagents were of analytical grade. All of the solutions used in this work were prepared using ultrapure water (>18.25 MΩ).2.2. Preparation of TWJ
To prepare the TWJ, we mixed two relevant single-stranded oligonucleotides (S1 and S2) in the hybridization buffer at the same molar ratios. The resulting mixture was then annealed via heating at 90 °C for 5 min, and gradually cooling down to room temperature. The hybridization buffer consisted of 20 mM Tris-Ac (pH 7.9), 50 mM KAc, 10 mM Mg(Ac)2 and 100 μg mL−1 BSA.2.3. Protein binding-protected DNA TWJ-mediated RCA process
Various concentrations of NF-κB p50 were incubated with the prepared TWJ (40 nM) in reaction buffer (20 mM Tris-Ac, pH 7.9, 50 mM KAc, 10 mM Mg(Ac)2, 100 μg mL−1 BSA) at 37 °C for 1.5 h to make the TF-DNA binding reaction to occur. Subsequently, we added 5.0 units of AlwI and the mixture reacted for additional 1.0 h at 37 °C to digest residual TWJ, then heating for 20 min at 75 °C to inactive AlwI. Next, 2 μL of 500 nM padlock probe, 3 μL 10 × T4 DNA ligase buffer (400 mM Tris–HCl, pH 7.8, 100 mM MgCl2, 100 mM DTT, 5 mM ATP), 1.0 unit T4 DNA ligase and water were added. We subsequently incubated the mixture at 37 °C for 40 min to allow the cyclization reaction to take place, then heating for 10 min at 75 °C to inactive T4 DNA ligase. Finally, 2.5 units phi29 DNA polymerase, 4 μL 10 × phi29 reaction buffer (330 mM Tris-Ac, pH 7.9, 100 mM Mg(Ac)2, 660 mM KAc, 1% Tween 20, 10 mM DTT), 3 μL 10 mM dNTPs, and 2.75 μL water were added and it was incubated for 60 min at 37 °C to allow the amplification reaction to happen. And the reaction was terminated by incubating for 10 min at 75 °C. For assaying the inhibition effect of inhibitor, the prepared TWJ (40 nM) was incubated with 75 nM NF-κB and various concentrations of oridonin, then operating as per the above-mentioned procedures.2.4. Fluorescence measurement and gel electrophoresis
After the RCA reaction, 5 μL NMM (37.5 μM) and 5 μL KCl (1.0 M) were added and the resulting mixture was incubated at 37 °C for 0.5 h. The fluorescence was measured via F-7000 spectrometer (Hitachi, Japan). The excitation wavelength and emission wavelength was 399 nm, 612 nm, respectively. The voltage of PMT detector was 700 V.The 15% polyacrylamide gel electrophoresis (PAGE) was performed in 1 × TBE buffer (89 mM Tris, 89 mM boric acid, 2.0 mM EDTA, pH 8.3). We then stained the gel via ethidium bromide. It was subsequently photographed with UV imaging system (Bio-RAD Laboratories Inc. USA).
3. Results and discussion
3.1. Principle of the TFs assay
The protocol of TFs detection based on protein binding-protected TWJ-mediated RCA is shown in Scheme 1. The TWJ contained three sections: target protein binding site (in blue), AlwI recognition site (in red) and sutured primer of RCA (in pink). The binding of target protein occupied the cleavage site of AlwI, four and five bases downstream the AlwI recognition site. And the great steric hindrance produced by protein-DNA binding protected TWJ from cleavage by AlwI. The protected TWJ with sutured primer of RCA was then annealed to the padlock probe. Both ends of padlock probe were specifically ligated in the presence of T4 ligase and the circular template was formed. Then, with the addition of phi29 DNA polymerase, the RCA reaction was initiated. In this case, the sutured primer was extended with a long G4 sequence, a copy of the circular template. Finally, the G4 sequence interacted with NMM to yield a great fluorescence enhancement. Residual TWJs were digested efficiently by AlwI. It was worthing mentioning that the Tm between cleaved two toeholds was designed to be lower than 15 °C under the reaction condition to insure their hybrid unstable,27 breaking up into two dissociative chains. Importantly, although the two dissociative chains both had some complementary bases with the padlock probe, the cyclization reaction could not happen because the 5′ tail and 3′ tail of the padlock probe are difficult to get close to each other without the intact sutured primer. The cleavage of AlwI thus effectively blocked RCA reaction and suppressed nonspecific amplification. | ||
| Scheme 1 Schematic illustration of the fluorescence assay for the sensitive and specific detection of TF. In TWJ, the blue segment, the red segment denote target protein binding site, AlwI recognition site, respectively. The cleavage site of AlwI is marked by red arrows. The two pink branches in TWJ is the sutural primer of RCA. | ||
3.2. Feasibility research
The successful formation of TWJ and effective cleavage of probe by AlwI played decisive role in the method proposed here. PAGE (15%) was employed to verify the two processes, as shown in Fig. 1A. Compared to lane 2 and lane 3, the top of lane 4 showed a obvious band, indicating the hybrid product TWJ was obtained. After the AlwI treatment, TWJ was digested into bands with higher mobility, verifying the effective release of chains under the action of AlwI.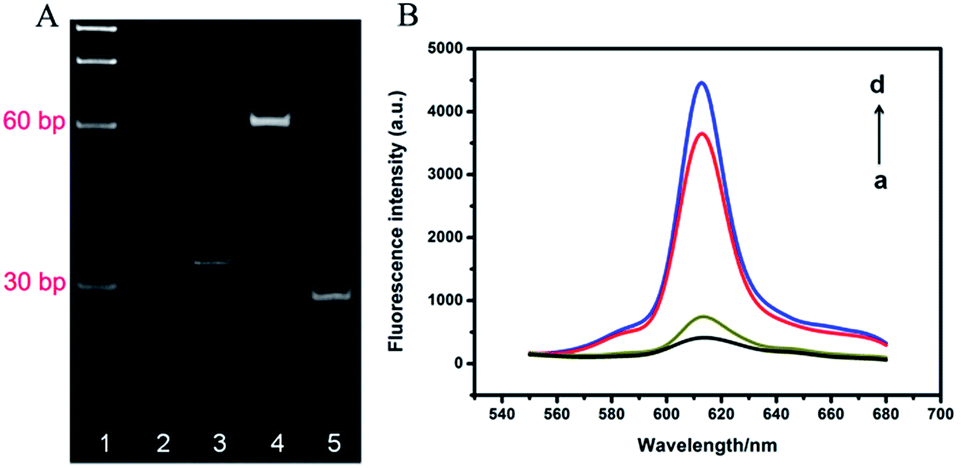 | ||
| Fig. 1 (A) The PAGE analysis of the formation of TWJ and probe cleavage of AlwI. Lane 1: marker, lane 2: S1, lane 3: S2, lane 4: S1 + S2, lane 5: S1 + S2 + AlwI. (B) Fluorescence emission spectra under different conditions (all contained 1.0 U T4 DNA ligase + 2.5 U phi29 DNA polymerase + 0.75 mM dNTPs + 3.75 μM NMM): (a) 20 nM padlock probe, (b) 40 nM TWJ + 5.0 U AlwI + 20 nM padlock probe (negative), (c) 40 nM TWJ + 20 nM NF-κB p50 + 5.0 U AlwI + 20 nM padlock probe (positive), (d) 20 nM TWJ + 20 nM padlock probe. | ||
To verify the feasibility of protein binding-protected TWJ-mediated RCA process, fluorescent imaging under different conditions was also performed, as shown in Fig. 1B. The padlock probe showed a weak fluorescence intensity (curve a), and only a little fluorescent enhancement appeared after adding TWJ and AlwI (curve b), demonstrating TWJ was efficiently digested by AlwI and the product could not initiate RCA. Nevertheless, the fluorescence intensity significantly increased in the presence of NF-κB p50, TWJ and AlwI (curve c), indicating the TWJ cleavage by AlwI can be effectively hindered via the specific binding of NF-κB p50 and RCA reaction was successfully performed. Furthermore, the fluorescence intensity of curve c was only slightly lower than the intensity of curve d, which represented the specimen containing intact TWJ and padlock probe. This result also indicated the RCA reaction was successfully performed.
3.3. Optimization of the probe design
In the proposed strategy, the hybridization between TWJ and padlock probe was a key step to generate an amplified signal. Thus, we firstly investigated the hybridization mode between them, as shown in Fig. 2A. Three different TWJs (TWJ 1, TWJ 2, and TWJ 3) were prepared by different S1 and S2 (Table S1†), among which the 3′ tail of S2 had different matched nucleotide bases with 5′ tail of padlock probe. Fig. 2B indicated that the hybridization between TWJ 3 and padlock probe exhibited the largest fluorescence intensity, which was almost equal to the intensity produced by entire primer and padlock probe. The possible reason is the S1–S2 double helix with about 2 nm diameter made the distance between the two ends of padlock probe too large for cyclization reaction in TWJ 1. Whereas the distance became close enough to allow the cyclization reaction to happen in TWJ 3, because of the joint action of the 5′ tail of S1 and three matched nucleotide bases of the 3′ tail of S2 to the 5′ tail of padlock probe.28 Thus TWJ 3 was chosen to perform the followed work. | ||
| Fig. 2 (A) Different hybridization modes between TWJ and padlock probe: (1) TWJ 1, (2) TWJ 2, (3) TWJ 3. (B) Fluorescence emission spectra under different hybridization modes between TWJ and padlock probe: (a) padlock probe, (b) TWJ 1 + padlock probe, (c) TWJ 2 + padlock probe, (d) TWJ 3 + padlock probe, (e) entire primer + padlock probe. | ||
3.4. Optimization of the reaction conditions
Reaction conditions had important impacts on the results of an assay. Therefore, we investigated the reaction conditions in order to obtain the best performance, using the value of ΔF as standard (ΔF = F − F0, F and F0 were the fluorescence intensity of the samples in the absence and presence of NF-κB p50, respectively).AlwI–TWJ interaction converted the assay of target protein to the DNA detection. To ensure the accuracy of this assay, the dosage of AlwI was investigated (Fig. S1†). It was observed that the value of ΔF gradually increased with the increasing dosage of AlwI, and attained a plateau at 5.0 U of AlwI. Thus 5.0 U of AlwI was chosen for subsequent experiments.
The concentrations of phi29 DNA polymerase and dNTPs were also optimized (Fig. S2 and S3†). 2.5 U of phi29 DNA polymerase and 0.75 mM dNTPs were eventually selected as the optimal concentration based on the experimental results. In addition, we also optimized the concentration of NMM (Fig. S4†). It was observed that value of ΔF reached a maximum with 3.75 μM of NMM. Thereby, 3.75 μM NMM was chosen as the optimum concentration.
3.5. Analytical performance of the assay
We next investigated the analytical performance of the proposed method with various NF-κB p50 concentrations under the optimal conditions. The result in Fig. 3A showed that the fluorescence intensity increased gradually with the increasing concentration of NF-κB p50 from 0 to 15 nM, demonstrating the close relationship between the cleavage activity of AlwI and the concentration of target protein. We obtained a good linearity within the range from 8 pM to 15 nM (Fig. 3B). The linear function for the concentrations of NF-κB p50 was ΔF = 9428.02 + 840.01![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) log10C (C is the concentrations of NF-κB p50 (8 pM to 15 nM); R2 = 0.998) with a detection limit of 6.8 pM (3σ/slope). The high sensitivity of the proposed method was probably ascribed to the high amplification efficiency of TWJ-mediated RCA reaction and AlwI-assisted background reduction. We also investigated the analytical performance using SYBR Green I as the indicator of RCA product29,30 (Fig. S5†). The linear function for the concentrations of NF-κB p50 was ΔF = 9993.67 + 910.70log10C (C is the concentrations of NF-κB p50 (15 pM to 15 nM); R2 = 0.992) with a detection limit of 13.2 pM (3σ/slope), which was slightly higher than NMM method (6.8 pM). The result indicated that NMM was more applicable to our design. It may result from the relatively high background due to the intercalation of SYBR Green I to TWJ.
log10C (C is the concentrations of NF-κB p50 (8 pM to 15 nM); R2 = 0.998) with a detection limit of 6.8 pM (3σ/slope). The high sensitivity of the proposed method was probably ascribed to the high amplification efficiency of TWJ-mediated RCA reaction and AlwI-assisted background reduction. We also investigated the analytical performance using SYBR Green I as the indicator of RCA product29,30 (Fig. S5†). The linear function for the concentrations of NF-κB p50 was ΔF = 9993.67 + 910.70log10C (C is the concentrations of NF-κB p50 (15 pM to 15 nM); R2 = 0.992) with a detection limit of 13.2 pM (3σ/slope), which was slightly higher than NMM method (6.8 pM). The result indicated that NMM was more applicable to our design. It may result from the relatively high background due to the intercalation of SYBR Green I to TWJ.
 | ||
| Fig. 3 (A) Fluorescence curves obtained from different NF-κB p50 concentrations. (B) Linear relationship between the fluorescence enhancement and NF-κB p50 concentration. Error bars indicate the standard deviation of 3 measurements. | ||
To evaluate the detection specificity, the fluorescence responses towards NF-κB p50 against three other interfering proteins were studied. As shown in Fig. 4, only NF-κB p50 (15 nM) produced large fluorescence response, whereas three potential interfering proteins (150 nM) including CEA, c-Jun protein, FR and BSA gave low intensity. The experimental results clearly indicated the strategy we proposed here exhibited high specificity for the detection of NF-κB p50.
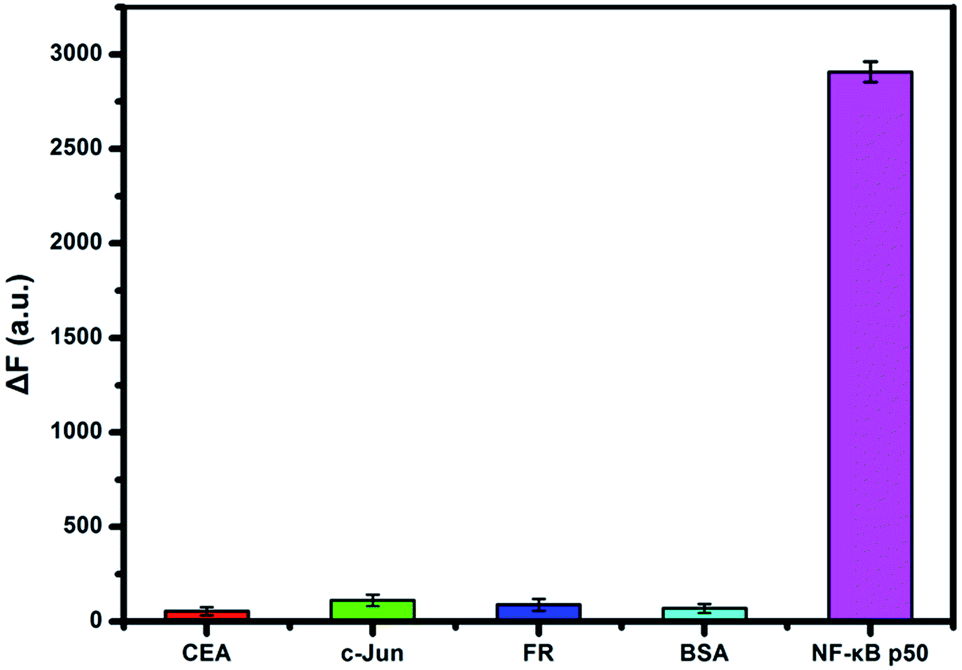 | ||
| Fig. 4 Specificity of the assay to NF-κB p50 in comparison to interfering proteins (CEA, c-Jun, FR and BSA). Experimental conditions: 15 nM NF-κB p50, 150 nM interfering proteins. Error bars indicate the standard deviation of 3 measurements. | ||
We also investigated the precision and reproducibility using the relative standard deviations (RSD) to evaluate the reliability of the proposed assay. The RSD obtained from the same batch were 4.3%, 3.5% and 1.8% at 25 pM, 500 pM and 5 nM target proteins. The RSD obtained from three different batches by measuring the same specimen in three days were 6.0%, 3.2% and 2.5%. The above results showed the satisfactory precision and reproducibility of the developed strategy.
3.6. Assaying the inhibition of NF-κB p50
The utility of the protein binding-protected TWJ-mediated RCA strategy for assaying the inhibition of NF-κB p50 was also investigated. Herein, we selected a model inhibition of NF-κB, oridonin, which can retard the binding between NF-κB p50 and DNA binding site (Fig. S6†).31 The relative fluorescence intensity showed gradually reduction with the increasing dose of oridonin, verifying the strategy can be applied to screen inhibitors of NF-κB p50. It is worthing mentioning that this also further demonstrate that the cleavage activity of AlwI was closely associated with the protein-DNA binding.3.7. Feasibility in complex biological matrices
The 10% HeLa cells lysate were spiked with NF-κB p50 at three concentrations (50 pM, 500 pM and 5 nM). Table S2† illustrated that the obtained recoveries of the three samples were 96.0%, 102.7% and 98.7% with RSD of 2.1%, 4.1% and 2.3%. Compared with the reported methods in which the recoveries were from 92.5% to 107%,29,32 these results verified that the proposed method was reliable and had the potential for real sample application.3.8. Real sample analysis
To assess the practical application of the strategy proposed here, we further measured the NF-κB p50 in real sample, HeLa cell nuclear extracts. According to EMSA,33 the standard method of transcription factor detection, and the current works,34,35 we used the total protein content as a measurement criterion to monitor NF-κB activity. As shown in Fig. 5, the fluorescence intensity gradually increased with the increasing target concentration. We obtained a good linearity from 0.5 ng μL−1 to 40 ng μL−1. The linear function for the concentrations of HeLa cell nuclear extracts was ΔF = 102.14 + 66.76C (C is the concentrations of HeLa cell nuclear extracts (0.5–40 ng μL−1); R2 = 0.998) with a detection limit of 0.34 ng μL−1 (3σ/slope), which was superior or comparable to the previously reported works for NF-κB p50 detection.36 The results indicated that the strategy proposed here held great promise for practical application. | ||
| Fig. 5 (A) Fluorescence curves obtained from different concentrations of HeLa cell nuclear extracts. (B) Linear relationship between the fluorescence enhancement and the concentration of HeLa cell nuclear extracts. Error bars indicate the standard deviation of 3 measurements. | ||
4. Conclusions
Overall, we developed a label-free fluorescence strategy for sensitive and specific detection of NF-κB p50 based on protein binding-protected TWJ-mediated RCA reaction and AlwI-assisted background suppression. Compared to previously reported methods for TFs detection, the strategy is unique in the following features: (i) the trifunctional TWJ served together as NF-κB p50, AlwI recognition probe and sutured primer of RCA, avoiding the requirement of multiple probes design. (ii) Once residual TWJs were cleaved into two parts by AlwI, the RCA can be effectively blocked, thus suppressing nonspecific amplification. (iii) AlwI did not need a defined cleavage sequence, thus without requirement for binding site of target protein with a specific sequence for enzyme cleavage, enhancing the versatility of the assay. Sensitive detection of NF-κB p50 was achieved with a low detection limit of 6.8 pM and a broad detection range over 4 orders of magnitude. Moreover, NF-κB p50 in HeLa cell nuclear extracts was successfully measured with a low detection limit of 0.34 ng μL−1, and this strategy can be used to screen inhibitors of NF-κB. The results indicated that the proposed strategy held great promise for studying of TFs in disease diagnostics and drug developments.Acknowledgements
This work was supported by National Natural Sciences Foundation of China (Grant No. 21375078 and 21475077) and Project Funded by China Postdoctoral Science Foundation (2015M582074).Notes and references
- C. O. Pabo and R. T. Sauer, Annu. Rev. Biochem., 1992, 61, 1053–1095 CrossRef CAS PubMed.
- F. Spitz and E. E. Furlong, Nat. Rev. Genet., 2012, 13, 613–626 CrossRef CAS PubMed.
- M. Marin, A. Karis, P. Visser, F. Grosveld and S. Philipsen, Cell, 1997, 89, 619–628 CrossRef CAS PubMed.
- M. Boiani and H. R. Scholer, Nat. Rev. Mol. Cell Biol., 2005, 6, 872–884 CrossRef CAS PubMed.
- M. Eggert, A. Kluter, U. K. Zettl and G. Neeck, Curr. Pharm. Des., 2004, 10, 2787–2796 CrossRef CAS PubMed.
- K. L. Clark, K. E. Yutzey and D. W. Benson, Annu. Rev. Physiol., 2006, 68, 97–121 CrossRef CAS PubMed.
- A. P. Sanchez and K. Sharma, Expert Rev. Mol. Med., 2009, 11, e13 CrossRef PubMed.
- M. Karin, Nature, 2006, 441, 431–436 CrossRef CAS PubMed.
- A. J. Hampshire, D. A. Rusling, V. J. Broughton-Head and K. R. Fox, Methods, 2007, 42, 128–140 CrossRef CAS PubMed.
- M. Fried and D. M. Crothers, Nucleic Acids Res., 1981, 9, 6505–6525 CrossRef CAS PubMed.
- L. M. Hellman and M. G. Fried, Nat. Protoc., 2007, 2, 1849–1861 CrossRef CAS PubMed.
- S. V. Gupta, R. M. McGowen, D. M. Callewaert, T. R. Brown, Y. Li and F. H. Sarkar, J. Immunoassay Immunochem., 2005, 26, 125–143 CrossRef CAS.
- T. Heyduk and E. Heyduk, Nat. Biotechnol., 2002, 20, 171–176 CrossRef CAS PubMed.
- E. Knoll and T. Heyduk, Anal. Chem., 2004, 76, 1156–1164 CrossRef CAS PubMed.
- A. V. Belisle, A. J. Bonham, N. O. Reich, F. Ricci and K. W. Plaxco, J. Am. Chem. Soc., 2011, 133, 13836–13839 CrossRef PubMed.
- J. K. Wang, T. X. Li, X. Y. Guo and Z. H. Lu, Nucleic Acids Res., 2005, 33, e23 CrossRef PubMed.
- A. Cao and C. Y. Zhang, Anal. Chem., 2013, 85, 2543–2547 CrossRef CAS PubMed.
- X. F. Liu, L. Ouyang, X. H. Cai, Y. Q. Huang, X. M. Feng, Q. L. Fan and W. Huang, Biosens. Bioelectron., 2013, 41, 218–224 CrossRef CAS PubMed.
- D. L. Ma, T. Xu, D. S. H. Chan, B. Y. W. Man, W. F. Fong and C. H. Leung, Nucleic Acids Res., 2011, 39, e67 CrossRef CAS PubMed.
- H. J. He, R. Pires, T. N. Zhu, A. H. Zhou, A. K. Gaigalas, S. G. Zou and L. L. Wang, BioTechniques, 2007, 43, 93–98 CrossRef PubMed.
- L. J. Qu, P. Y. Jin, X. Chu, J. H. Jing and R. Q. Yu, Anal. Chem., 2010, 82, 6015–6024 CrossRef PubMed.
- Y. Xu, K. D. Lunnen and H. Kong, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 12990–12995 CrossRef CAS PubMed.
- V. N. Antipova, L. A. Zheleznaya and N. V. Zyrina, FEMS Microbiol. Lett., 2014, 357, 144–150 CAS.
- E. J. Cho, L. Yang, M. Levy and A. D. Ellington, J. Am. Chem. Soc., 2005, 127, 2022–2023 CrossRef CAS PubMed.
- S. S. Oh, K. Plakos, X. Lou, Y. Xiao and H. T. Soh, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 14053–14058 CrossRef CAS PubMed.
- D. Hu, F. Pu, Z. Huang, J. Ren and X. Qu, Chem.–Eur. J., 2010, 16, 2605–2610 CrossRef CAS PubMed.
- Y. Li, L. Liang and C. Y. Zhang, Anal. Chem., 2013, 85, 11174–11179 CrossRef CAS PubMed.
- W. L. Song, Q. Zhang and W. B. Sun, Chem. Commun., 2015, 51, 2392–2395 RSC.
- C. X. Li, X. Y. Qiu, Z. H. Hou and K. Q. Deng, Biosens. Bioelectron., 2015, 64, 505–510 CrossRef CAS PubMed.
- Y. T. Zhou, Q. Huang, J. M. Gao, J. X. Lu, X. Z. Shen and C. H. Fan, Nucleic Acids Res., 2010, 38, e156 CrossRef PubMed.
- C. H. Leung, S. P. Grill, W. Lam, Q. B. Han, H. D. Sun and Y. C. Cheng, Mol. Pharmacol., 2005, 68, 286–297 CAS.
- J. H. Chen, X. Zhang, S. X. Cai, D. Z. Wu, J. Lin, C. Y. Li and J. Zhang, Biosens. Bioelectron., 2014, 53, 12–17 CrossRef CAS PubMed.
- P. Renard, I. Ernest, A. Houbion, M. Art, H. L. Calvez, M. Raes and J. Remacle, Nucleic Acids Res., 2001, 29, e21 CrossRef CAS PubMed.
- Y. Zhang, J. Hu and C. Y. Zhang, Anal. Chem., 2012, 84, 9544–9549 CAS.
- F. Ma, Y. Yang and C. Y. Zhang, Anal. Chem., 2014, 86, 6006–6011 CrossRef CAS PubMed.
- D. S. Zhu, L. Wang, X. W. Xu and W. Jiang, Biosens. Bioelectron., 2016, 75, 155–160 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: DNA sequences and supporting figures. See DOI: 10.1039/c6ra12535b |
| This journal is © The Royal Society of Chemistry 2016 |