
Harnessing the pyrroloquinoxaline scaffold for FAAH and MAGL interaction: definition of the structural determinants for enzyme inhibition†
Abstract
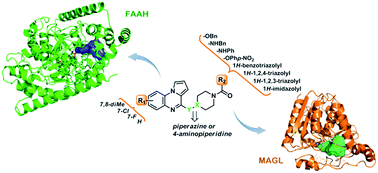
This paper describes the development of piperazine and 4-aminopiperidine carboxamides/carbamates supported on a pharmacogenic pyrroloquinoxaline scaffold as inhibitors of the endocannabinoid catabolizing enzymes fatty acid amide hydrolase (FAAH) and monoacylglycerol lipase (MAGL). Structure–activity relationships and molecular modelling studies allowed the definition of the structural requirements for dual FAAH/MAGL inhibition and led to the identification of a small set of derivatives (compounds 5e, i, k, m) displaying a balanced inhibitory profile against both enzymes, with compound 5m being the frontrunner of the subset. Favorable calculated physico-chemical properties suggest further investigation for specific analogues.
 Please wait while we load your content...
Please wait while we load your content...

