
Synthesis of 2-hydroxy-3-alkyl-2-phenyl-2,3-dihydroquinazolin-4(1H)-one via molybdenum hexacarbonyl mediated CO gas- and ligand free carbonylative reactions†
J. Subba Raoab,
A. Venkateswara Rao*b,
T. Krishnaa,
V. N. Murthya,
J. Rajesha and
A. Raghunadh*a
aTechnology Development Centre, Custom Pharmaceutical Services, Dr Reddy's Laboratories Ltd, Hyderabad 500049, India. E-mail: raghunadha@drreddys.com
bDepartment of Chemistry, Koneru Lakshmaiah University (KLU), Green Fields, Vaddeswaram, Guntur, Andhra Pradesh 522502, India
First published on 5th July 2016
Abstract
Carbon monoxide gas and ligand-free conditions were developed for the synthesis of 2-hydroxy-3-alkyl-2-phenyl-2,3-dihydroquinazolin-4(1H)-one via catalytic carbonylation with molybdenum hexacarbonyl as an efficient carbonylating agent for the three-component reaction of isatoic anhydride, amine, iodobenzene. Mo(CO)6 is a solid carbon monoxide source. The quinazolinone synthesis proceeds via a sequential series of reactions such as nucleophilic attack of the amine group on the carbonyl group of isatoic anhydride followed by ring opening, subsequent decarboxylation, carbonylation and heterocyclization.
The development of novel and efficient synthetic methods towards the building of a nitrogen containing heterocyclic ring is an important area of synthetic and medicinal chemistry as many drugs or bioactive agents belong to this class of heterocycles.1 2-Arylquinazolin-4(3H)-ones are a highly important class of heteroaromatic compounds that are widely found in pharmaceuticals, and bioactive molecules. The quinazoline core unit is found in many natural products, including alkaloids like bouchardatine 1,2 batracylin 2,3 ophiuroidine 3,4 (−)-benzomalvin A 4,5 luotonon A 5,6 luotonon B 6, luotonon E 7 and asperlicin D 8 (Fig. 1).7 Some quinazolinone based natural products e.g. febrifugine and isofebrifugine have been identified as potential antimalarial agents.8
 | ||
| Fig. 1 Selected examples of bioactive natural products which contain quinazolin-4(3H)-ones skeleton. | ||
Medicinal chemists have synthesized a variety of arylquinazolin-4(3H)-ones compounds with different biological activities by installing various active groups. Because of varied biological properties of quinazolinone derivatives, a number of methodologies have been developed for their synthesis towards quinazolin-4(3H)-ones derivatives. Rao et al. reported a versatile method for the solid-phase synthesis of differentially substituted quinazolin-4(3H)-ones.9 Recently, Besson et al. reported a ligand-free palladium catalyzed and copper-assisted intermolecular C-2–H arylations with (hetero)aryl iodides.10
Following our efforts on the functionalization of quinazolin-4(3H)-one derivatives, Robert et al. also reported the molybdenum-mediated synthesis of quinazolin-4(3H)-ones via cyclocarbonylation using microwave irradiation.11 In the context of our ongoing research work, recently our research group has demonstrated that substituted quinazolin-4(3H)-ones based biologically active natural products and their derivatives.12
Carbonylation is a classical synthetic methodology in organic chemistry for introducing carbon monoxide to C–C and C–N bond formation.13 Organometallic methodologies have been also examined as a substitute for phosgene chemistry. A variety of metal centres can be used as catalysts in the presence of CO2 or CO. However, not all transition-metal catalysts are useful in the oxidative carbonylation. Oxidative carbonylation of primary amines to substituted urea's has been reported for transition-metal catalysts involving Ni, Co, Mn, Ru and most commonly Pd.14
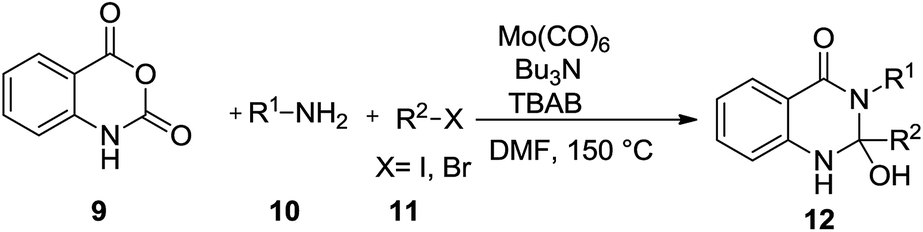
Bhanage reported palladium-catalyzed synthesis of primary amides by aminocarbonylation of aryl and heteroaryl iodides.15 Yamane reported a similar molybdenum-mediated carbamoylation of aryl halides under thermal conditions with molybdenum carbonyl complexes.16 Roberts and team also reported molybdenum-mediated carbonylation of aryl halides with nucleophiles to give carbonyl products under microwave irradiation.17 We have explored the possibility of an operatively simple and novel synthesis of 2-hydroxy-3-alkyl-2-phenyl-2,3-dihydroquinazolin-4(1H)-one derivatives 12 with isatoic anhydride 9,18 amine 10 and iodobenzene 11 with molybdenum hexacarbonyl mediated CO gas-free cyclocarbonylation19 via multi-component reaction strategy.
The retro synthetic strategy employed for the synthesis of 2-hydroxy-3-alkyl-2-phenyl-2,3-dihydroquinazolin-4(1H)-one derivatives is depicted in Scheme 1. The phenylquinazolin-4(3H)-one derivative 13 could be obtained via dehydration of 12 under heating. Initially when isatoic anhydride 9, amine 10a and iodobenzene 11a were treated under the conditions applied for construction of dihydroquinazolin-4(1H)-one in DMF using 0.2 eq. of Mo(CO)6 gives 23% of the desired product 12a.
 | ||
| Scheme 1 Retrosynthesis of 12. | ||
In an effort to develop optimal conditions, various reaction parameters like different catalysts, bases and solvents were studied for the preparation of 12. The CO sources, namely Mo(CO)6, Cr(CO)6 and W(CO)6 were screened (Table 1, entries 1–5). The best result was obtained when the reaction was performed in the presence of 1.0 eq. of Mo(CO)6. Further various solvents like DMF, 1,4-dioxane, DMSO, and diglyme were screened (Table 1, entries 5–8) finally it was found that DMF was the suitable solvent for the carbonylation reaction. Once we established the suitable CO source and solvent for the synthesis of 2-hydroxy-3-alkyl-2-phenyl-2,3-dihydroquinazolin-4(1H)-ones, further we screened various bases like triethylamine, tributylamine, potassium carbonate and cesium carbonate in DMF (Table 1, entries 9–12). The best result was obtained when the reaction was performed with tributylamine as a base. However, the reaction did not give the corresponding carbonylation product when the reaction was conducted with potassium carbonate as well as with cesium carbonate, we screened various temperatures at lower temperature (below 100 °C) product formation was not observed only isatoic anhydride open product 14 was observed. Further we screened TBAB and TBAI both are working well and we observed 3% less yield with TBAI.
| Entry | CO source (eq.) | Solvent | Base | Isolated yield (%) |
|---|---|---|---|---|
| a Reaction and conditions: isatoic anhydride (1.0 eq.), n-butyl amine (1.0 eq.), 4-iodo-1,1′-biphenyl (1.0 eq.), Mo(CO)6 (1.0 eq.) and Bu3N (1.2 eq.), TBAB (0.2 eq.), in DMF at 150 °C. | ||||
| 1 | Mo(CO)6 (0.2) | DMF | Bu3N | 23 |
| 2 | Cr(CO)6 (0.2) | DMF | Bu3N | 8 |
| 3 | W(CO)6 (0.2) | DMF | Bu3N | 11 |
| 4 | Mo(CO)6 (0.5) | DMF | Bu3N | 45 |
| 5 | Mo(CO)6 (1.0) | DMF | Bu3N | 72 |
| 6 | Mo(CO)6 (1.0) | 1,4-Dioxane | Bu3N | 31 |
| 7 | Mo(CO)6 (1.0) | DMSO | Bu3N | 42 |
| 8 | Mo(CO)6 (1.0) | Diglyme | Bu3N | 36 |
| 9 | Mo(CO)6 (1.0) | DMF | TEA | 10 |
| 10 | Mo(CO)6 (1.0) | DMF | K2CO3 | 0 |
| 11 | Mo(CO)6 (1.0) | DMF | Cs2CO3 | 0 |
| 12 | Without CO source | DMF | Cs2CO3 | 0 |
With these conditions in hand, the scope of this transformation was tested using several substituted aliphatic and aromatic amine and various aryl halides (Table 2). When the reaction was conducted with aryl iodide high yields were obtained when compared with aryl bromide.
|
||||
|---|---|---|---|---|
| Entry | Amine 1; R1= | Aryl 2; R2= | Product 3 | Yielda (%) |
| a Isolated yields. | ||||
| 1 | 10a; n-hexyl | 11a; 4-biphenyl |  |
78 |
| 2 | 10b; –CH2Ph | 11a |  |
72 |
| 3 | 10c; –CH2C6H4OMe-p | 11a | 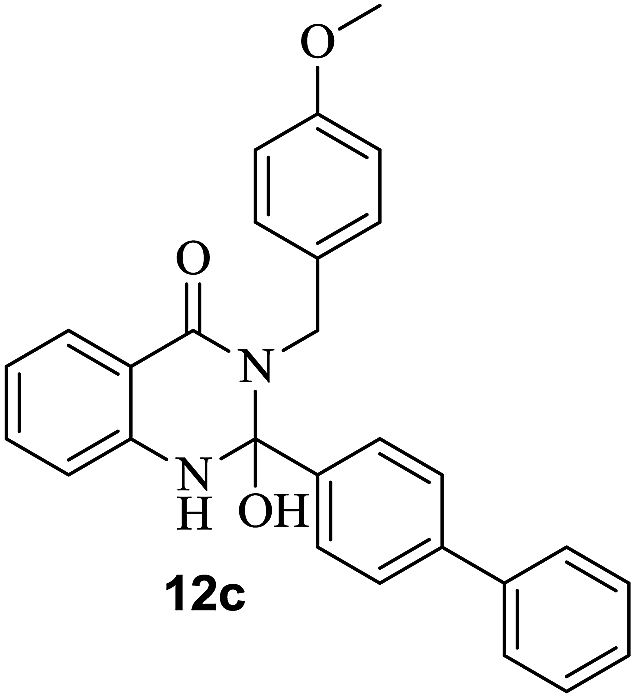 |
62 |
| 4 | 10d; –cycloheptyl | 11a |  |
75 |
| 5 | 10e; –(S)-1-phenylethyl | 11a |  |
67 |
| 6 | 10a | 11b; bromobenzene | 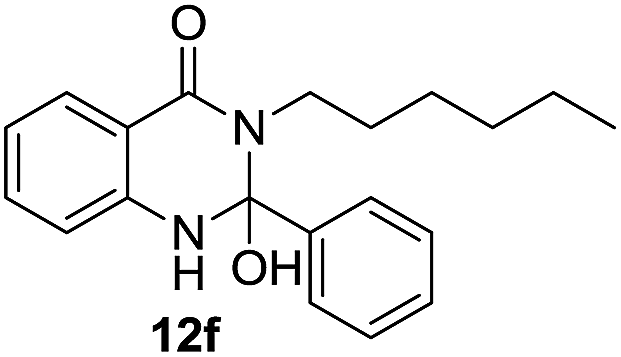 |
71 |
| 7 | 10b | 11b |  |
72 |
| 8 | 10d | 11b |  |
70 |
| 9 | 10e | 11b |  |
63 |
| 10 | 10a | 11c; BrC6H4OMe-p |  |
58 |
| 11 | 10b | 11c | 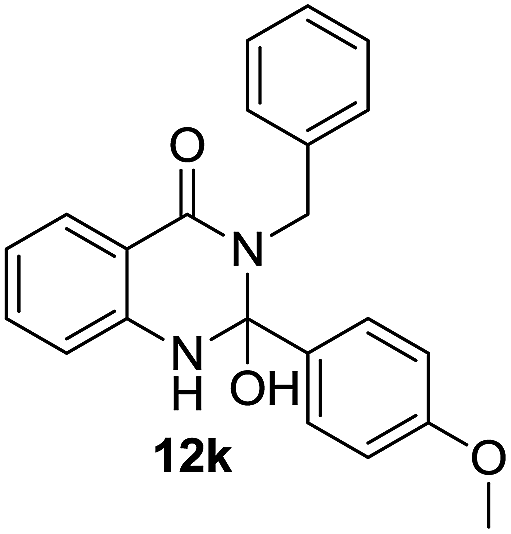 |
56 |
| 12 | 10d; | 11c | 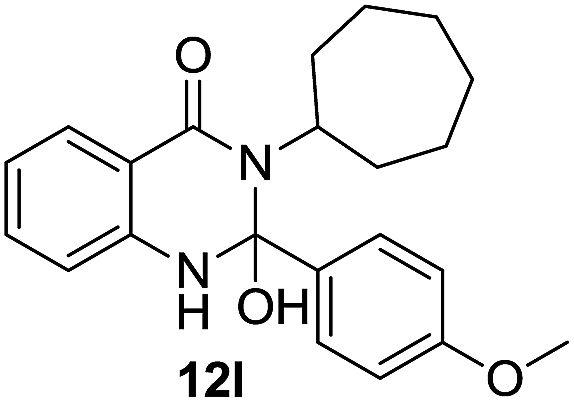 |
57 |
| 13 | 10a | 11d; BrC6H4F-p |  |
36 |
| 14 | 10a | 11e; 2-iodothiophene |  |
85 |
| 15 | 10e | 11e |  |
82 |
| 16 | 10a | 11f; 1-bromo-3-methoxybenzene |  |
62 |
| 17 | 10b | 11f |  |
55 |
| 18 | 10b | 11g; 1-bromo-2-methoxybenzene |  |
45 |
Less yield was observed when the reaction was conducted with 4-fluro bromo benzene (entry 13). Placing electron withdrawing groups in the para position seems to be less reactivity. During the reaction around 5% aromatized product 13 formation was observed, when we maintained reaction for 48 h at 150 °C product 12 was completely undergoing for the de hydration leading to the formation of 13 with 75% of isolated yield.
The Scheme 2 represents a plausible mechanism for the three component reaction leading to the compound 12. The nucleophilic attack of primary amine on carbonyl group of isatoic anhydride followed by ring opening and subsequent decarboxylation provided the compound 14·Et4NBr readily displaces a CO ligand from Mo(CO)6 to give Mo(CO)5Br·NEt4 and this complex reacts readily with nitrogen nucleophile of 14 will yield the 15, which on deprotonation provides 16. This could then undergo oxidative-addition or CO insertion to give 18 or 17. Intermediate 17 undergo oxidative-addition with aryl halide to give 19. Alternatively intermediate 18 undergo CO insertion to give 19 and 20. Reductive elimination of 19 and 20 would give the diamide 21 and subsequent cyclization of 21 would give the product 12, which undergo the de hydration leading to the formation of 13.
 | ||
| Scheme 2 The proposed reaction mechanism for the formation of 12. | ||
In conclusion, we have developed a short and efficient novel methodology for the synthesis of 2-hydroxy-3-alkyl-2-phenyl-2,3-dihydroquinazolin-4(1H)-one derivative via multi-component reaction strategy in good yields from isatoic anhydride, amine and iodobenzene in a one pot process. Carbon monoxide gas and ligand free condition was developed for the synthesis of 2-hydroxy-3-alkyl-2-phenyl-2,3-dihydroquinazolin-4(1H)-one via catalytic carbonylation with molybdenum hexacarbonyl as an efficient carbonylating agent for three-component reaction of isatoic anhydride, amine, iodobenzene and Mo(CO)6 as a solid carbon monoxide source. The quinazolinone synthesis proceeds via a sequential series of reactions such as nucleophilic attack of amine group on carbonyl group of isatoic anhydride followed ring opening and subsequent decarboxylation, carbonylation and heterocyclization.
Acknowledgements
The authors would like to thank Dr Vilas Dahanukar, Dr H. Rama Mohan, Dr K. B. Shiva Kumar and the analytical group of CPS-DRL for spectral data.Notes and references
- Bioactive Heterocyclic Compound Classes: Pharmaceuticals and Agrochemicals, ed. C. Lamberth and J. Dinges, Wiley-VCH, New York, 2012 Search PubMed.
- S. B. Mhaske and N. P. Argade, J. Org. Chem., 2004, 69, 4563 CrossRef CAS PubMed.
- (a) K. Dzierzbicka, P. Trzonkowski, P. L. Sewerynek and A. Mysliwski, J. Med. Chem., 2003, 46, 978 CrossRef CAS PubMed; (b) J. Guillaumel, S. Léonce, A. Pierre, P. Renard, B. Pfeiffer, P. B. Arimondo and C. Monneret, Eur. J. Med. Chem., 2006, 41, 379 CrossRef CAS PubMed; (c) R. Yilin, C. Yun Feng, C. Ting, A. Y. Chen, C. Yu, L. F. Liu and C. C. Cheng, Pharm. Res., 1993, 10, 918 CrossRef.
- N. K. Utkina and S. A. Fedoreev, Tetrahedron Lett., 2007, 48, 4445 CrossRef CAS.
- (a) T. Sugimori, T. Okawa, S. Eguchi, A. Kakehi, E. Yashima and Y. Okamoto, Tetrahedron, 1998, 54, 7997 CrossRef CAS; (b) H. H. Sun, C. J. Barrow, D. M. Sedlock, A. M. Gillum and R. Cooper, J. Antibiot., 1994, 47, 515 CrossRef CAS PubMed.
- (a) T. Harayama, Y. Morikami, Y. Shigeta, H. Abe and Y. Takeuchi, Synlett, 2003, 847 CrossRef CAS; (b) Z. Ma, Y. Hano, T. Nomura and Y. Chen, Bioorg. Med. Chem. Lett., 2004, 14, 1193 CrossRef CAS PubMed; (c) M. C. Tseng, Y. W. Chu, H. P. Tsai, C. M. Lin, J. Hwang and Y. H. Chu, Org. Lett., 2011, 13, 920 CrossRef CAS PubMed; (d) S. P. Chavan and R. Sivappa, Tetrahedron, 2004, 60, 9931 CrossRef CAS; (e) M. B. Wagh, R. Shankar, U. K. S. Kumar and C. H. Gill, Synlett, 2011, 84 CAS; (f) L. Nagarapu, H. K. Gaikwad and R. Bantu, Synlett, 2012, 23, 1775 CrossRef CAS.
- (a) F. He, B. M. Foxman and B. B. Snider, J. Am. Chem. Soc., 1998, 120, 6417 CrossRef CAS; (b) T. Sugimori, T. Okawa, S. Eguchi, A. Kakehi, E. Yashima and Y. Okamoto, Tetrahedron, 1998, 54, 7997 CrossRef CAS.
- (a) Y. Takeuchi, K. Azuma, K. Takakura, H. Abe, H.-S. Kim, Y. Wataya and T. Harayama, Tetrahedron, 2001, 57, 1213 CrossRef CAS; (b) T. Taniguchi and K. Ogasawara, Org. Lett., 2000, 2, 3193 CrossRef CAS PubMed; (c) M. Sugiura, H. Hagio, R. Hirabayashi and S. Kobayashi, J. Am. Chem. Soc., 2001, 123, 12510 CrossRef CAS PubMed; (d) Y. Tekeuchi, H. Abe and T. Harayama, Chem. Pharm. Bull., 1999, 47, 905 CrossRef; (e) S. Kobayashi, M. Ueno, R. Suzuki, H. Ishitani, H.-S. Kim and Y. Wataya, J. Org. Chem., 1999, 64, 6833 CrossRef CAS PubMed.
- A. V. D. Rao, B. P. Vykunteswararao, T. Bhaskarkumar, R. J. Nivrutti, D. Kalita, J. D. L. kumar, V. Siddaiah, S. Paul Douglas and A. Raghunadh, Tetrahedron Lett., 2015, 56, 4714 CrossRef.
- (a) A. S. Golubev, V. O. Bogomolov, A. F. Shidlovskii, L. G. Dezhenkova, A. S. Peregudov, A. A. Shtil and N. D. Chkanikova, Russ. Chem. Bull., 2010, 59, 209 CrossRef CAS; (b) K. Nacro, C. Zha, P. R. Guzzo, R. J. Herr, D. Peace and T. D. Friedrich, Bioorg. Med. Chem., 2007, 15, 4237 CrossRef CAS PubMed; (c) T. Harayama, A. Hori, G. Serban, Y. Morikami, T. Matsumoto, H. Abe and Y. Takeuchi, Tetrahedron, 2004, 60, 10645 CrossRef CAS; (d) T. Harayama, Y. Morikami, Y. Shigeta, H. Abe and Y. Takeuchi, Synlett, 2003, 847 CrossRef CAS; (e) S. Laclef, M. Harari, J. Godeau, I. Schmitz-Afonso, L. Bischoff, C. Hoarau, V. Levacher, C. Fruit and T. Besson, Org. Lett., 2015, 17, 1700–1703 CrossRef CAS PubMed.
- B. Roberts, D. Liptrot, T. Luker, M. J. Stocks, C. Barber, N. Webb and R. Dods, Tetrahedron Lett., 2011, 52, 3793 CrossRef CAS.
- (a) K. R. Raghavendra, M. Ramamohan, A. Raghunadh, M. Suresh Babu, S. Praveen Kumar, D. Kalita, E. Laxminarayana, B. Prasad and M. Pal, RSC Adv., 2015, 5, 61575 RSC; (b) V. N. Murthy, S. P. Nikumbh, S. Praveen Kumar, L. Vaikunta Rao and A. Raghunadh, Tetrahedron Lett., 2015, 56, 5767 CrossRef.
- (a) J. Wannberg and M. Larhed, in Modern Carbonylation Methods, ed. L. Kollar, Wiley-VCH, Weinheim, 2008, pp. 93 Search PubMed; (b) S. R. Borhade, A. Sandstrom and P. I. Arvidsson, Org. Lett., 2013, 15, 1056 CrossRef CAS PubMed; (c) P. Nordeman, L. R. Odell and M. Larhed, J. Org. Chem., 2012, 77, 11393 CrossRef CAS PubMed; (d) A. Begouin and M.-J. R. P. Queiroz, Eur. J. Org. Chem., 2009, 2820 CrossRef CAS; (e) D. Liptrot, L. Alcaraz and B. Roberts, Adv. Synth. Catal., 2010, 352, 2183 CrossRef CAS; (f) D. N. Sawant, Y. S. Wagh, K. D. Bhatte and B. M. Bhanage, J. Org. Chem., 2011, 76, 5489 CrossRef CAS PubMed.
- (a) M. Beller, B. Cornils, C. D. Frohning and C. W. Kohlpaintner, J. Mol. Catal. A: Chem., 1995, 104, 17 CrossRef CAS; (b) R. Skolda-Foldes and L. Kollar, Curr. Org. Chem., 2002, 6, 1097 CrossRef; (c) C. F. J. Barnard, Organometallics, 2008, 27, 5402 CrossRef CAS; (d) Modern Carbonylation Methods, ed. Kollár L., Wiley-VCH, Weinheim, 2008 Search PubMed; (e) A. Brennführer, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2009, 48, 4114 CrossRef PubMed; (f) R. Grigg and S. P. Mutton, Tetrahedron, 2010, 66, 5515 CrossRef CAS; (g) X.-F. Wu, H. Neumann and M. Beller, Chem. Soc. Rev., 2011, 40, 4986 RSC.
- (a) P. J. Tambade, Y. P. Patil, M. J. Bhanushali and B. M. Bhanage, Tetrahedron Lett., 2008, 49, 2221 CrossRef CAS; (b) P. J. Tambade, Y. P. Patil, M. J. Bhanushali and B. M. Bhanage, Synthesis, 2008, 15, 2347 Search PubMed.
- W. Ren and M. Yamane, J. Org. Chem., 2010, 75, 8410 CrossRef CAS PubMed.
- B. Roberts, D. Liptrot, L. Alcaraz, T. Luker and M. J. Stocks, Org. Lett., 2010, 12, 4280 CrossRef CAS PubMed.
- M. G. A. Shvekhgeimer, Chem. Heterocycl. Compd., 2001, 37, 385 CrossRef CAS.
- (a) O. Belda, N.-F. Kaiser, U. Bremberg, M. Larhed, A. Hallberg and C. Moberg, J. Org. Chem., 2000, 65, 5868 CrossRef CAS PubMed; (b) N.-F. K. Kaiser, U. Bremberg, M. Larhed, C. Moberg and A. Hallberg, Angew. Chem., Int. Ed., 2000, 39, 3595 CrossRef; (c) N. F. K. Kaiser, A. Hallberg and M. Larhed, J. Comb. Chem., 2002, 4, 109 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, spectral data for all new compounds, copies of spectra. See DOI: 10.1039/c6ra12510g |
| This journal is © The Royal Society of Chemistry 2016 |