
Functionalised dihydroazo pyrimidine derivatives from Morita–Baylis–Hillman acetates: synthesis and studies against acetylcholinesterase as its inhibitors†
Abstract
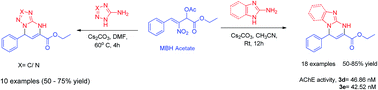
A wide array of dihydro[1,5]azo[1,2-a]pyrimidine 2-esters have been synthesised at room temperature in moderate to good yields through one-pot reaction between nitrostyrene derived MBH acetates and aminoazole derivatives with γ-α cyclisation. The reaction involves a cascade SN2 reaction followed by intramolecular Michael addition–cyclisation with the generation of two new carbon–nitrogen bonds. The structure was further confirmed by single X-ray crystal analysis. Docking and in vitro studies of dihydrobenzimidazo pyrimidine derivatives against acetylcholinesterase (AChE) have been performed and the compounds 3d and 3e exhibit potent inhibitory activities with an IC50 of 46.8 nM and 42.5 nM respectively.
 Please wait while we load your content...
Please wait while we load your content...

