
Ru(II)-p-cymene complexes containing esters of chiral D/L-phenylalanine derived aroylthiourea ligands for enantioselective reduction of pro-chiral ketones†
Mani Mary Sheebaa,
Manoharan Muthu Tamizhb,
Sundaram Ganesh Babua,
Nattamai S. P. Bhuvaneshc and
Ramasamy Karvembu*a
aDepartment of Chemistry, National Institute of Technology, Tiruchirappalli 620015, India. E-mail: kar@nitt.edu; Fax: +91 431 2500133; Tel: +91 431 2503631
bDepartment of Chemistry, Siddha Central Research Institute (SCRI), Arignar Anna Govt. Hospital Campus, Arumbakkam, Chennai-600106, India
cDepartment of Chemistry, Texas A & M University, College Station, TX 77842, USA
First published on 11th July 2016
Abstract
A series of new chiral aroylthiourea ligands was derived from unprotected D/L-phenylalanine: (R)/(S)-2-(3-benzoylthioureido)-3-phenylpropanoic acid (L1/L2), (R)/(S)-2-(3-(thiophene-2-carbonyl)thioureido)-3-phenylpropanoic acid (L3/L4) and (R)/(S)-2-(3-(furan-2-carbonyl)thioureido)-3-phenylpropanoic acid (L5/L6). Chiral Ru(II) complexes (1–6) were obtained from the reactions between the chiral ligands (L1–L6) and [RuCl2(p-cymene)2]2 through in situ catalytic esterification of the ligand in the presence of methanol solvent. The ligands and complexes were characterized by analytical and spectral (1H NMR, 13C NMR, Mass, FT-IR, electronic) techniques. The molecular structure of the ligand L1 showed the presence of an unprotected acid group and that of the representative complexes confirmed the conversion of acid to ester. The X-ray structure of two of the complexes (3 and 6) revealed the sulfur only monodentate coordination of the aroylthiourea ligands. All the chiral complexes turned out to be efficient catalysts for the enantioselective reduction of aromatic pro-chiral ketones in the presence of 2-propanol and NaOH to produce chiral alcohols in excellent conversions (up to 99%) and enantiomeric excesses (up to 99%) within 10–12 h.
1 Introduction
α-Amino acids are one of the readily obtainable and inexpensive natural chiral compounds, and are used as chiral auxiliaries or reagents for the synthesis of optically active molecules.1 Although aroylthiourea ligands prepared from primary and secondary amines have been widely used to synthesize transition metal complexes for various applications,2–7 amino acid derived aroylthiourea ligands which might provide more coordination possibilities, are not yet known. It was expected that the introduction of an amino acid moiety into the aroylthiourea ligand would make aroylthiourea complexes soluble in water. These factors induced us to synthesize chiral aroylthiourea ligands from D/L-phenylalanine.The chemistry of amino acid complexes of transition metals has received considerable attention.8–10 Amino acids/amino acid derived ligands and their complexes were used as catalysts in enantioselective reactions such as asymmetric transfer hydrogenation (ATH),11–16 epoxidation,17,18 deamination,19 aldol reaction,20 silylation of alcohols,21 and allylic alkylation.22 The number of Ru-p-cymene complexes with amino acids/amino acid derived ligands explored as catalysts for ATH is relatively less but they showed an outstanding activity.11–16 The amino acids/amino acid derived ligands which employed with Ru-p-cymene for ATH of ketones are L-prolinamide,23 tert-butyl-((S)-1-(((S)-2-hydroxypropyl)amino)-1-oxopropan-2-yl)carbamate, tert-butyl((S)-1-(((R)-2-hydroxy-1-phenylethyl)amino)-1-thioxopropan-2-yl)carbamate,24 (S)-2-amino-3-methyl-N-phenylbutanamide, (S)-2-amino-N-(4-methoxyphenyl)-3-methyl butanamide,14 S-phenylalanine,25 (R)-N-hydroxypyrrolidine-2-carboxamide,11 tert-butyl-((S)-1-(((S)-2-hydroxypropyl)amino)-1-oxopropan-2-yl)carbamate,15 (2S)-N-(1-(1H-benzo[d]imidazol-2-yl)ethyl)pyrrolidine-2-carboxamide,26 tert-butyl-N-((1S)-2-{[(1R)-2-hydroxy-1-phenylethyl]amino}-1-methyl-2-oxoethyl)carbamate16 and tert-butyl-((S)-1-(((S)-2-hydroxy-2-phenylethyl)amino)-1-oxo-3-phenylpropan-2-yl)carbamate.27
Ru(II)-p-cymene complexes were reported as highly active catalysts or pre-catalysts for various organic transformations.28 [RuCl2(p-cymene)]2 itself acted as a catalyst in many organic transformations.29,30 It was observed in the present work that the amino acids were converted into corresponding esters in the presence of [RuCl2(p-cymene)]2 and methanol prior to their coordination with Ru(II). The esters derived from amino acids are very important intermediates in organic synthesis and effective anticancer and antiviral drugs. The amino esters have been used as prodrugs to increase oral bioavailability in pharmaceuticals and helped to improve the physicochemical properties and reduce the toxicity.31 Amino acid esters have wide applications in the area of peptide synthesis,32 polymer synthesis,33,34 asymmetric synthesis35–38 and medicinal chemistry.39,40
Chirality is significant for the designing of drug molecules. The pharmacological effect of organic compounds is very often caused by only one enantiomer while the other one can have a different effect or even have no effect. For that reason, direct methods of obtaining optically pure chiral compounds are highly in demand. Conventional organic reactions almost always lead to a racemic mixture, but asymmetric reactions can give a single enantiomeric product. The asymmetric reduction of pro-chiral ketones under mild catalytic hydrogen transfer conditions will offer a highly attractive route for the formation of enantiomerically pure secondary alcohols, an important building blocks, and synthetic intermediates in organic synthesis and pharmaceutical industry41–43 and an important class of fine chemicals.44
We report here the synthesis and characterization of new chiral aroylthiourea ligands derived from D/L-phenylalanine such as (R)/(S)-2-(3-benzoylthioureido)-3-phenylpropanoic acid (L1/L2), (R)/(S)-2-(3-(thiophene-2-carbonyl)thioureido)-3-phenyl-propanoic acid (L3/L4) and (R)/(S)-2-(3-(furan-2-carbonyl)thioureido)-3-phenylpropanoic acid (L5/L6), and their ester Ru-p-cymene complexes (1–6). These Ru-p-cymene complexes were successfully applied as catalysts for the enantioselective reduction of aromatic ketones to their corresponding enantiomerically enriched secondary alcohols. The in situ transformation of amino acid to amino ester in the presence of [RuCl2(p-cymene)]2 as a catalyst was confirmed.
2 Results and discussion
2.1 Synthesis of the ligands and complexes
The chiral ligands (L1–L6) were synthesized from benzoyl chloride or thiophene-2-carbonyl chloride or furan-2-carbonyl chloride, potassium thiocyanate and D-phenylalanine or L-phenylalanine in acetone–ethanol mixture (Scheme 1). The chiral Ru(II) complexes (1–6) were obtained from the reactions between [RuCl2(η6-p-cymene)]2 and the chiral ligands (L) in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of toluene and methanol (Scheme 2). Methanol is essential for the solubility of the ligands. Due to the presence of [RuCl2(η6-p-cymene)]2 and methanol, the unprotected amino acid was converted into amino ester before coordination. This conversion clearly indicated that [RuCl2(p-cymene)2]2 acted not only as a Ru precursor but also as a catalyst for the in situ production of amino ester. To confirm this, amino acid (L1) was stirred at 27 °C in the presence of catalytic amount of [RuCl2(η6-p-cymene)]2 (Scheme 3). The formation of ester was attested by 1H NMR analysis. This conversion is interesting because amino esters are very important for the design of many drug molecules and pro-drugs. However esterification of the ligands prevented the solubility of their Ru-p-cymene complexes in water. The ligands and Ru-p-cymene complexes were characterized by elemental analysis, and UV-Vis, FT-IR, 1H NMR, 13C NMR and HR-MS spectroscopic methods. The molecular structure of the ligand L1 and complexes (3 and 6) was confirmed by single crystal X-ray crystallography. The optical rotation value of the compounds was obtained by polarimetric studies. All the ligands and complexes were air stable and soluble in toluene, benzene, CH3OH, C2H5OH, CHCl3, CH2Cl2, CH3CN, DMF and DMSO.
:1 mixture of toluene and methanol (Scheme 2). Methanol is essential for the solubility of the ligands. Due to the presence of [RuCl2(η6-p-cymene)]2 and methanol, the unprotected amino acid was converted into amino ester before coordination. This conversion clearly indicated that [RuCl2(p-cymene)2]2 acted not only as a Ru precursor but also as a catalyst for the in situ production of amino ester. To confirm this, amino acid (L1) was stirred at 27 °C in the presence of catalytic amount of [RuCl2(η6-p-cymene)]2 (Scheme 3). The formation of ester was attested by 1H NMR analysis. This conversion is interesting because amino esters are very important for the design of many drug molecules and pro-drugs. However esterification of the ligands prevented the solubility of their Ru-p-cymene complexes in water. The ligands and Ru-p-cymene complexes were characterized by elemental analysis, and UV-Vis, FT-IR, 1H NMR, 13C NMR and HR-MS spectroscopic methods. The molecular structure of the ligand L1 and complexes (3 and 6) was confirmed by single crystal X-ray crystallography. The optical rotation value of the compounds was obtained by polarimetric studies. All the ligands and complexes were air stable and soluble in toluene, benzene, CH3OH, C2H5OH, CHCl3, CH2Cl2, CH3CN, DMF and DMSO.
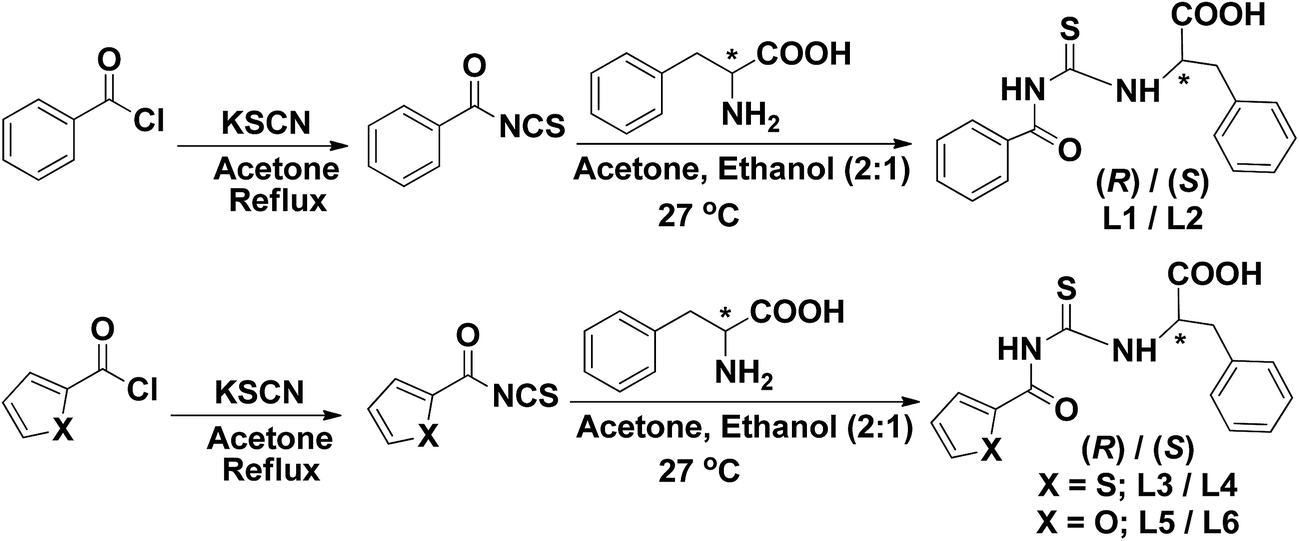 | ||
| Scheme 1 Synthesis of ligands (L1–L6). | ||
 | ||
| Scheme 2 Synthesis of complexes (1–6). | ||
 | ||
| Scheme 3 Conversion of amino acid to amino ester. | ||
2.2 Characterization of the ligands and complexes
In the 1H NMR spectra of all the ligands (L1–L6), the signal due to carboxyl O–H proton was found as a broad singlet in the region 13.29–13.31 ppm. The thiocarbonyl attached N–H, and carbonyl and thiocarbonyl attached N–H protons were observed as doublet at 10.97–11.23 ppm and singlet at 11.27–11.59 ppm respectively. The resonances owing to the aromatic ring (phenyl, thiophene or furan) protons were appeared at 6.72–8.35 ppm in the spectra of all the ligands. A multiplet was observed at 5.09–5.16 ppm for the proton present in the chiral carbon. The CH2 protons of the ligands were observed as two doublet of doublets at 3.16–3.35 ppm. In the 1H NMR spectra of all the complexes (1–6), the signal due to carboxyl O–H proton was disappeared and a new signal observed at 2.59–3.91 ppm indicated the conversion of acid to ester. The new signals seemed in the region 1.26–1.34, 2.19–3.30 and 5.21–5.47 ppm indicated the presence of p-cymene moiety in the complexes.5 All other chemical shift values of the complexes were almost similar to those of the corresponding free ligands. The 13C NMR spectra of the ligands showed signals at 36.0 and 58.6 ppm for the CH2 and asymmetric carbons respectively. The signals appearing at 112.6–147.8 ppm in the spectra of all the ligands were assigned to the aromatic (phenyl, thiophene or furan ring) carbons. The resonances due to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and CS were observed around 158.3–168.8 and 171.2–175.5 ppm respectively. The carboxyl carbon signal was observed at 179.9–180.3 ppm.13C NMR spectra of the complexes also confirmed the acid to ester conversion. The ester COO and CH3 carbon signals were observed at 178.7–179.6 and 51.9–52.7 ppm respectively. All other chemical shift values did not undergo significant change upon coordination of the ligand to Ru. The new signals observed around 18.3, 22.2, 30.4, 82.6, 82.7, 84.2, 84.3, 100.0 and 103.4 ppm confirmed the presence of p-cymene in all the complexes.5
O and CS were observed around 158.3–168.8 and 171.2–175.5 ppm respectively. The carboxyl carbon signal was observed at 179.9–180.3 ppm.13C NMR spectra of the complexes also confirmed the acid to ester conversion. The ester COO and CH3 carbon signals were observed at 178.7–179.6 and 51.9–52.7 ppm respectively. All other chemical shift values did not undergo significant change upon coordination of the ligand to Ru. The new signals observed around 18.3, 22.2, 30.4, 82.6, 82.7, 84.2, 84.3, 100.0 and 103.4 ppm confirmed the presence of p-cymene in all the complexes.5
Further, the formation of the complexes was established from the d–d (441–448 nm) and charge transfer transitions (328–339 nm) observed in the UV-visible spectra of the complexes. The FT-IR spectra of the complexes were compared with that of the free ligands. The frequencies of the CO and amide N–H stretching modes were almost unaltered upon coordination, but the CS stretching frequency of the complexes decreased from the region 1240–1246 to 1183–1196 cm−1, which strongly suggested that the ligands were bound to the Ru ion via the sulfur atom only.3,5 The carboxylic acid CO stretching frequency of the ligands observed in the region 1705–1718 cm−1 was increased to 1738–1741 cm−1 in the complexes, which confirmed the conversion of acid to ester prior to complexation.45 Coordination mode of the ligands and functional group transformation were unambiguously confirmed by the single crystal X-ray diffraction study.
2.3 X-ray crystallographic study
The structure of the ligand L1 and complexes 3 and 6 was elucidated by single crystal X-ray diffraction study and are shown in the Fig. 1–3. The ligand and complexes crystallized in the chiral orthorhombic crystal system with the space group P212121. The molecular structure of ligand L1 evidently displayed the presence of acid group in the free ligand. The thermal ellipsoidal plot of 3 and 6 confirmed the monodentate sulfur coordination of the thiourea ligand in the complexes and the formation of ester group in the coordinated ligand. In the complexes Ru ion displayed half-sandwich “3-legged piano-stool” coordination geometry. The hydrogen bonding interactions N(1)–H(1)⋯O(3) and N(2)–H(2)⋯Cl(1) characteristic of this type of complexes were clearly seen3,5 (Fig. 2 and 3). | ||
| Fig. 1 Thermal ellipsoidal plot of L1 showing the atomic labeling scheme and thermal ellipsoids at the 50% probability level. Selected bond distances (Å) and angles (°): S(1)–C(8) 1.683(4), O(1)–C(1) 1.227(5), O(2)–C(10) 1.203(5), O(3)–H(3) 0.8400, O(3)–C(10) 1.325(5), N(1)–H(1) 0.8800, N(2)–H(2) 0.8800, C(8)–N(1)–H(1) 116.4, C(8)–N(2)–H(2) 119.0, N(2)–C(8)–S(1) 123.2(3), N(1)–C(8)–S(1) 119.4(3). | ||
 | ||
| Fig. 2 Thermal ellipsoidal plot of 3 showing the atomic labeling scheme and thermal ellipsoids at the 50% probability level. Selected bond distances (Å) and angles (°): Ru(1)–S(1) 2.4157(9), Ru(1)–Cl(1) 2.4247(10), Ru(1)–Cl(2) 2.4239(10), S(1)–Ru(1)–Cl(1) 90.91(3), S(1)–Ru(1)–Cl(2) 91.36(4), Cl(2)–Ru(1)–Cl(1) 86.38(3), C(1)–S(1)–Ru(1) 116.00(11). | ||
 | ||
| Fig. 3 Thermal ellipsoidal plot of 6 showing the atomic labeling scheme and thermal ellipsoids at the 50% probability level. Selected bond distances (Å) and angles (°): Ru(1)–S(1) 2.4221(11), Ru(1)–Cl(1) 2.4275(11), Ru(1)–Cl(2) 2.4280(10), S(1)–Ru(1)–Cl(1) 90.54(4), S(1)–Ru(1)–Cl(2) 91.40(4), Cl(2)–Ru(1)–Cl(1) 86.90(3), C(1)–S(1)–Ru(1) 115.60(13). | ||
2.4 Enantioselective reduction of ketones
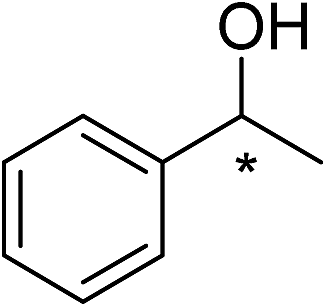
The chiral Ru-p-cymene complexes (1–6) acted as effective catalysts for the asymmetric reduction of pro-chiral ketones to their corresponding secondary chiral alcohols. The reactions were carried out in the presence of NaOH and 2-propanol at 82 °C by using the optimized substrate:NaOH:catalyst ratio of 200:200:1. The conversions of up to 99% and enantiomeric excesses (ee) of up to 99% were achieved within 10 h for acetophenone and 12 h for other substituted ketones. The conversion and ee values were determined by GC and chiral HPLC analyses respectively and are given in Table 1.
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | Substrate | Product | Conversionb (%) | ee (%)c/configurationd | TONe |
| a Reactions were carried out at 82 °C using 1 mmol of ketone, 0.005 mmol of Ru(II) complex in 5 mL of 2-propanol and 1 mmol of NaOH for 10–12 h.b The conversion was determined by GC-MS or GC.c ee was determined by chiral HPLC.d Absolute configuration was determined from the optical rotation value.e TON = moles of the product formed/moles of the catalyst used. | ||||||
| 1 | (1) |  |
 |
99 | 98/R | 198 |
| 2 | (2) | 99 | 95/R | 198 |
||
| 3 | (3) | 99 | 54/S | 198 |
||
| 4 | (4) | 99 | 58/R | 198 |
||
| 5 | (5) | 99 | 62/S | 198 |
||
| 6 | (6) | 99 | 60/S | 198 |
||
| 7 | (1) |  |
 |
74 | 52/R | 148 |
| 8 | (2) | 77 | 88/R | 154 |
||
| 9 | (3) | 96 | 92/R | 192 |
||
| 10 | (4) | 80 | 88/R | 160 |
||
| 11 | (5) | 90 | 99/R | 180 |
||
| 12 | (6) | 76 | 92/R | 152 |
||
| 13 | (1) |  |
 |
98 | 99/R | 196 |
| 14 | (2) | 91 | 99/R | 182 |
||
| 15 | (3) | 96 | 99/R | 192 |
||
| 16 | (4) | 85 | 99/R | 170 |
||
| 17 | (5) | 95 | 74/R | 190 |
||
| 18 | (6) | 94 | 95/R | 188 |
||
| 19 | (1) |  |
 |
93 | 94/R | 186 |
| 20 | (2) | 83 | 62/R | 166 |
||
| 21 | (3) | 63 | 85/R | 126 |
||
| 22 | (4) | 97 | 80/R | 194 |
||
| 23 | (5) | 84 | 48/R | 168 |
||
| 24 | (6) | 94 | 80/R | 188 |
||
| 25 | (1) |  |
 |
99 | 70/R | 198 |
| 26 | (2) | 99 | 89/R | 198 |
||
| 27 | (3) | 99 | 99/R | 198 |
||
| 28 | (4) | 99 | 99/R | 198 |
||
| 29 | (5) | 99 | 91/R | 198 |
||
| 30 | (6) | 99 | 99/R | 198 |
||
| 31 | (1) |  |
 |
85 | 99/S | 170 |
| 32 | (2) | 89 | 97/S | 178 |
||
| 33 | (3) | 77 | 99/S | 154 |
||
| 34 | (4) | 83 | 98/S | 166 |
||
| 35 | (5) | 83 | 98/S | 166 |
||
| 36 | (6) | 78 | 99/S | 156 |
||
| 37 | (1) |  |
 |
77 | 99/R | 154 |
| 38 | (2) | 99 | 99/R | 198 |
||
| 39 | (3) | 98 | 99/R | 196 |
||
| 40 | (4) | 99 | 99/R | 198 |
||
| 41 | (5) | 99 | 95/S | 198 |
||
| 42 | (6) | 99 | 99/R | 198 |
||

The potency of the present catalytic system was compared with the previously existing catalysts based on amino acids with [RuCl2(η6-p-cymene)]2, mainly with respect to the enantioselective reduction of acetophenone (Scheme 4). Enantioselective reduction of ketones with (S)-pyrrolidine-2-carboxamide-Ru-p-cymene catalytic system showed reasonably good conversion (90%) and enantiomeric excess (79%) in 20 h.23 But in the present catalytic system, catalyst 1 showed superior catalytic activity of 99% conversion and 98% ee within 10 h. [RuCl2(p-cymene)]2 with tert-butyl-((S)-1-(((R)-2-hydroxy-1-phenylethyl)amino)-1-thioxopropan-2-yl)carbamate ligand provided only 35% conversion and 20% ee,24 which was inferior to catalysts 1–6. Nevertheless, [RuCl2(p-cymene)]2 with tert-butyl-((S)-1-(((R)-2-hydroxy-1-phenylethyl)amino)-1-oxopropan-2-yl)carbamate ligand gave 83% conversion and 92% ee,24 in which ee is comparable with that provided by 1 and 2. (S)-2-Amino-3-methyl-N-phenylbutanamide/(S)-2-amino-N-(4-methoxyphenyl)-3-methylbutanamide with Ru-p-cymene offered the conversion of 77/98% and ee of 34/33%, and the highest ee reported in this system was only 47%14 which was significantly lower than our present results. Ru(II)-p-cymene complexes containing (S)-phenylalanine were used as catalysts in ATH of ketones in isopropanol/KOH medium and the reported conversion was 74% and ee was only 28%.25 But in the present case Ru-p-cymene with phenylalanine derived aroylthiourea exhibited an excellent conversion (99%) and ee (98%). (R)-N-Hydroxypyrrolidine-2-carboxamide/[RuCl2(p-cymene)]2 catalytic system resulted 6% conversion and 41% ee after 17 h,11 which is poor compared to the present results. Ru-p-cymene/tert-butyl-((S)-1-(((S)-2-hydroxypropyl)amino)-1-oxopropan-2-yl)carbamate system furnished 94% conversion and 96% ee15 which are comparable with the performance of 1 and 2. The ligand tert-butyl-((S)-1-(((S)-2-hydroxy-2-phenylethyl)amino)-1-oxo-3-phenylpropan-2-yl)carbamate on combination with [RuCl2(p-cymene)]2 showed 86% conversion and 95% ee;27 the ee is comparable with that exhibited by 1 and 2. [RuCl2(p-cymene)]2 bearing tert-butyl-((S)-1-(((R)-2-hydroxy-1-phenylethyl)amino)-1-oxopropan-2-yl)carbamate demonstrated a superior result (95% conversion and 93% ee) within 5 h.16 (2S)-N-(1-(1H-Benzo[d]imidazol-2-yl)ethyl)pyrrolidine-2-carboxamide-Ru-p-cymene catalyst displayed an inferior result (65% ee).26 The efficiency of the present catalysts was compared with our previous Ru-p-cymene catalysts containing aroylthiourea derived from (R)/(S)-1-phenylethylamine. The present catalysts which contain amino acid derived aroylthiourea were more active as it took only 10–12 h for the quantitative conversion of ketones to their respective chiral alcohols with 99% ee; but the same reactions previously required 24 h for completion.5
 | ||
| Scheme 4 Efficacy of reported amino acids/amino acid derived ligands with [RuCl2(η6-p-cymene)]2 towards asymmetric reduction of acetophenone. | ||
2-Methyl acetophenone was converted into 1-(o-tolyl)ethanol with up to 96% conversion and 99% ee (Table 1, entries 7–12). All the catalysts selectively yielded R-alcohols. Enantiopure fluoro-organic compounds are rare and have various biological applications in the fields of anti-cancer, anti-viral and anti-infection.46–48 The half-sandwich Ru-p-cymene complexes (1–6) were used as catalysts for the conversion of pro-chiral 4-fluoro acetophenone to its corresponding chiral secondary alcohol with the conversions and ee up to 99% (Table 1, entries 13–18). Even though many ketones were enantioselectively reduced by ATH, reduction of heterocyclic ketones was rarely presented. The asymmetric reduction of 2-acetylfuran was achieved in our method which gave corresponding chiral alcohol with 93% conversion and 94% ee using catalyst 1 (Table 1, entries 19–24). The reduction of 2-acetylpyridine was performed, which provided corresponding chiral alcohol with 99% conversion and 99% ee using catalysts 3, 4 and 6 (Table 1, entries 25–30). All the catalysts (1–6) were selectively offered the R alcohol. Chiral benzhydrol is widely used in the synthesis of various pharmaceuticals.49 Using the present catalytic system, conversion of 4-methoxy benzophenone and 4-bromo benzophenone to corresponding chiral benzhydrol was accomplished (Table 1, entries 30–42 respectively). Based on our previous experience, arene might control the enantioselectivity and change of arene might lead to different enantioselectivity, which is under investigation.
3 Experimental section
3.1 General methods
The solvents were dried and stored over activated molecular sieves. [RuCl2(η6-p-cymene)]2 was prepared by following a literature procedure.50 UV-visible spectra were recorded using a Shimadzu 2600 spectrophotometer, operating in the range of 200–800 nm. FT-IR spectra were recorded in the 4000–600 cm−1 region on a Nicolet iS5 FT-IR spectrophotometer. CHNS analyses were performed using a Perkin Elmer 2400 series II elemental analyzer. 1H and 13C NMR spectra were recorded on a Bruker 500 MHz and 125 MHz spectrometer, respectively. Melting points were determined in open capillary tubes on a Sigma melting point apparatus and are uncorrected. Catalysis experiments were monitored using a Shimadzu GCMS-QP 2010 Ultra gas chromatograph mass spectrometer or Shimadzu GC 2010 gas chromatograph equipped with a Restek-5 capillary column. ee values were determined using a Shimadzu HPLC instrument with a Daicel Chiralcel OB-H column. Specific rotation values were measured on a Rudolph Autopol IV polarimeter.3.2 Synthesis of ligands (L1–L6)
A solution of benzoyl chloride (0.6 mL, 5 mmol)/thiophene-2-carbonyl chloride (0.5 mL, 5 mmol)/furan-2-carbonyl chloride (0.5 mL, 5 mmol) in acetone (30 mL) was added to a suspension of potassium thiocyanate (0.4859 g, 5 mmol) in acetone (30 mL). The reaction mixture was heated (70 °C) under reflux for 45 minutes and then cooled to room temperature. A solution of D/L-phenylalanine (0.8259 g, 5 mmol) in acetone (40 mL) and ethanol (20 mL) was added, and the resulting mixture was stirred for 12 h at 27 °C. Hydrochloric acid (0.1 N, 300 mL) was then added, and the resulting solid was filtered off. The solid product was washed with water and purified by recrystallization from an ethanol–dichloromethane mixture (1:2).
S attached N–H), 11.52 (s, 1H, CO and CS attached N–H), 13.31 (bs, 1H, COOH). 13C NMR (125 MHz, DMSO-d6): δ 36.0 (CH2), 58.6 (asymmetric carbon), 126.9, 128.4, 128.4, 128.6, 129.2, 131.9, 133.1, 136.2 (aromatic CH), 168.4 (CO), 171.3 (CS), 180.3 (COOH). FT-IR (KBr, cm−1): 3272 (m; ν(amide N–H)), 3196 (s; ν(thiourea N–H)), 1665 (s; ν(CO)), 1709 (s; ν(COOH)), 1240 (s; ν(CS)).S attached N–H), 11.51 (s, 1H, CO and CS attached N–H), 13.30 (bs, 1H, COOH). 13C NMR (125 MHz, DMSO-d6): δ 36.1 (CH2), 58.6 (asymmetric carbon), 126.9, 128.3, 128.3, 128.5, 129.1, 131.9, 133.0, 136.1 (aromatic CH), 168.3 (CO), 171.3 (CS), 180.2 (COOH). FT-IR (KBr, cm−1): 3272 (m; ν(amide N–H)), 3196 (s; ν(thiourea N–H)), 1665 (s; ν(CO)), 1709 (s; ν(COOH)), 1240 (s; ν(CS)).S attached N–H), 11.58 (s, 1H, CO and CS attached N–H), 13.30 (bs, 1H, COOH). 13C NMR (125 MHz, DMSO-d6): δ 36.0 (CH2), 58.5 (asymmetric carbon), 126.8, 128.3, 128.6, 129.1, 132.6, 135.2, 136.1, 136.4 (aromatic CH), 162.1 (CO), 171.2 (CS), 179.9 (COOH). FT-IR (KBr, cm−1): 3206 (m; ν(amide N–H)), 3168 (s; ν(thiourea N–H)), 1651 (s; ν(CO)), 1706 (s; ν(COOH)), 1246 (s; ν(CS)).S attached N–H), 11.59 (s, 1H, CO and CS attached N–H), 13.31 (bs, 1H, COOH). 13C NMR (125 MHz, DMSO-d6): δ 36.0 (CH2), 58.6 (asymmetric carbon), 126.8, 128.4, 128.7, 129.2, 132.6, 135.3, 136.2, 136.4 (aromatic CH), 162.2 (CO), 171.3 (CS), 179.9 (COOH). FT-IR (KBr, cm−1): 3205 (m; ν(amide N–H)), 3168 (s; ν(thiourea N–H)), 1651 (s; ν(CO)), 1705 (s; ν(COOH)), 1246 (s; ν(CS)).S attached N–H), 11.27 (s, 1H, CO and CS attached N–H), 13.29 (bs, 1H, COOH). 13C NMR (125 MHz, DMSO-d6): δ 36.0 (CH2), 58.6 (asymmetric carbon), 112.6, 118.6, 126.8, 128.4, 129.2, 136.1, 144.4, 148.4 (CH), 157.8 (CO), 171.3 (CS), 179.9 (COOH). FT-IR (KBr, cm−1): 3220 (m; ν(amide N–H)), 3162 (s; ν(thiourea N–H)), 1664 (s; ν(CO)), 1718 (s; ν(COOH)), 1245 (s; ν(CS)).S attached N–H), 11.27 (s, 1H, CO and CS attached N–H), 13.31 (bs, 1H, COOH). 13C NMR (125 MHz, DMSO-d6): δ 36.0 (CH2), 58.6 (asymmetric carbon), 112.6, 118.6, 126.9, 128.4, 129.2, 136.1, 144.4, 148.4 (CH), 157.8 (CO), 171.3 (CS), 179.9 (COOH). FT-IR (KBr, cm−1): 3220 (m; ν(amide N–H)), 3162 (s; ν(thiourea N–H)), 1664 (s; ν(CO)), 1717 (s; ν(COOH)), 1245 (s; ν(CS)).3.3 Synthesis of complexes (1–6)
[RuCl2(η6-p-cymene)]2 (122.5 mg, 0.2 mmol) and (R)/(S)-2-(3-benzoylthioureido)-3-phenylpropanoic acid (131 mg, 0.4 mmol) or (R)/(S)-2-(3-(thiophene-2-carbonyl)thioureido)-3-phenyl-propanoic acid (133.3 mg, 0.4 mmol) or (R)/(S)-2-(3-(furan-2-carbonyl)thioureido)-3-phenylpropanoic acid (127 mg, 0.4 mmol) were dissolved in toluene–methanol mixture (1:1) and stirred for 6 h at 27 °C. The solution was concentrated to 2 mL under reduced pressure, and addition of hexane (10–15 mL) gave a clear orange solid. The product was collected by filtration, washed with hexane and dried in vacuo.
O and CS attached N–H), 11.73 (d, 1H, J = 5 Hz, CS attached N–H). 13C NMR (125 MHz, CDCl3): δ 18.3 (CH3 of p-cymene), 22.2 (2CH3 of p-cymene), 30.5 (CH of p-cymene), 37.1 (CH2), 52.4 (ester CH3), 59.3 (asymmetric carbon), 82.5–84.3 (aromatic carbons of p-cymene), 99.9 and 103.5 (quaternary carbons of p-cymene), 127.2, 127.8, 128.5, 128.6, 128.7, 128.9, 129.2, 129.6, 131.1, 133.5, 135.8 (aromatic CH of the ligand), 168.5 (CO), 171.3 (CS), 179.4 (COOCH3). FT-IR (KBr, cm−1): 3220 (m; ν(amide N–H)), 3148 (s; ν(thiourea N–H)), 1674 (s; ν(CO)), 1740 (s; ν(COOCH3)), 1183 (s; ν(CS)). UV-vis (CHCl3; λ, nm ε, dm3 mol−1 cm−1): 441 (4584), 332 (19201), 253 (88187).O and CS attached N–H), 11.72 (d, 1H, J = 5 Hz, CS attached N–H). 13C NMR (125 MHz, CDCl3): δ 18.3 (CH3 of p-cymene), 22.2 (2CH3 of p-cymene), 30.5 (CH of p-cymene), 37.2 (CH2 of the ligand), 52.2 (ester CH3 of the ligand), 59.1 (asymmetric carbon), 82.5–84.3 (aromatic carbons of p-cymene), 99.9 and 103.5 (quaternary carbons of p-cymene), 127.3, 127.8, 128.5, 128.6, 128.7, 128.9, 129.2, 129.6, 131.0, 133.5, 135.2 (aromatic CH of the ligand), 168.6 (CO), 171.3 (CS), 179.1 (COOCH3). FT-IR (KBr, cm−1): 3217 (m; ν(amide N–H)), 3153 (s; ν(thiourea N–H)), 1673 (s; ν(CO)), 1741 (s; ν(COOCH3)), 1184 (s; ν(CS)). UV-vis (CHCl3; λ, nm ε, dm3 mol−1 cm−1): 448 (5635), 328 (24719), 253 (115366).O and CS attached N–H), 11.43 (d, 1H, J = 5 Hz, CS attached N–H). 13C NMR (125 MHz, CDCl3): δ 18.3 (CH3 of p-cymene), 22.2 (2CH3 of p-cymene), 30.4 (CH of p-cymene), 37.8 (CH2 of the ligand), 52.7 (ester CH3 of the ligand), 59.4 (asymmetric carbon), 82.6–84.3 (aromatic carbons of p-cymene), 100.0 and 103.4 (quaternary carbons of p-cymene), 127.5, 128.8, 129.2, 134.4, 134.7, 135.3, 135.9, (aromatic CH of the ligand), 162.9 (CO), 169.6 (CS), 179.9 (COOCH3). FT-IR (KBr, cm−1): 3211 (m; ν(amide N–H)), 3151 (s; ν(thiourea N–H)), 1660 (s; ν(CO)), 1740 (s; ν(COOCH3)), 1187 (s; ν(CS)). UV-vis (CHCl3; λ, nm ε, dm3 mol−1 cm−1): 444 (6288), 334 (30756), 294 (69381), 255 (95360).O and CS attached N–H), 11.43 (d, 1H, J = 10 Hz, CS attached N–H). 13C NMR (125 MHz, CDCl3): δ 18.3 (CH3 of p-cymene), 22.2–22.3 (2CH3 of p-cymene), 30.5 (CH of p-cymene), 37.0 (CH2), 52.5 (ester CH3 of the ligand), 59.4 (asymmetric carbon), 82.4–84.4 (aromatic carbons of p-cymene), 100.6 and 103.5 (quaternary carbons of p-cymene), 128.6, 128.7, 129.2, 129.6, 131.3, 134.3, 135.1, 136.2, (aromatic CH of the ligand), 162.5 (CO), 171.2 (CS), 178.6 (COOCH3). FT-IR (KBr, cm−1): 3207 (m; ν(amide N–H)), 3149 (s; ν(thiourea N–H)), 1660 (s; ν(CO)), 1740 (s; ν(COOCH3)), 1187 (s; ν(CS)). UV-vis (CHCl3; λ, nm ε, dm3 mol−1 cm−1): 446 (6735), 339 (32371), 294 (73127), 255 (100824).O and CS attached N–H), 11.29 (d, 1H, J = 10 Hz, CS attached N–H). 13C NMR (125 MHz, CDCl3): δ 18.4 (CH3 of p-cymene), 22.2–22.3 (2CH3 of p-cymene), 30.5 (CH of p-cymene), 38.0 (CH2 of the ligand), 52.4 (ester CH3 of the ligand), 59.3 (asymmetric carbon), 82.4–84.3 (aromatic carbons of p-cymene), 100.0 and 103.6 (quaternary carbons of p-cymene), 112.9, 118.0, 121.2, 127.2, 128.7, 129.2, 129.6, 135.7, 146.4, 147.7 (aromatic CH of the ligand), 158.3 (CO), 172.6 (CS), 179.5 (COOCH3). FT-IR (KBr, cm−1): 3222 (m; ν(amide N–H)), 3151 (s; ν(thiourea N–H)), 1682 (s; ν(CO)), 1738 (s; ν(COOCH3)), 1196 (s; ν(CS)). UV-vis (CHCl3; λ, nm ε, dm3 mol−1 cm−1): 439 (6884), 325 (30383), 286 (86958), 249 (71554).O and CS attached N–H), 11.22 (d, 1H, J = 5 Hz, CS attached N–H). 13C NMR (125 MHz, CDCl3): δ 18.3 (CH3 of p-cymene), 22.2 (2CH3 of p-cymene), 30.5 (CH of p-cymene), 37.1 (CH2 of the ligand), 52.7 (ester CH3 of the ligand), 59.1 (asymmetric carbon), 82.4–84.3 (aromatic carbons of p-cymene), 100.0 and 103.7 (quaternary carbons of p-cymene), 112.7, 121.3, 127.4, 128.7, 129.2, 129.5, 135.0, 144.6, 147.7, (aromatic CH of the ligand), 158.1 (CO), 171.4 (CS), 179.0 (COOCH3). FT-IR (KBr, cm−1): 3226 (m; ν(amide N–H)), 3150 (s; ν(thiourea N–H)), 1682 (s; ν(CO)), 1739 (s; ν(COOCH3)), 1196 (s; ν(CS)). UV-vis (CHCl3; λ, nm ε, dm3 mol−1 cm−1): 444 (6506), 325 (30005), 286 (86958), 251 (72342).3.4 X-ray structure determinations
Details of data collection are given in Table S1 (ESI†). The crystal was placed in a cold nitrogen stream maintained at 150 K. A BRUKER APEX2 and APEX3 X-ray (three-circle) diffractometer was employed for crystal screening, unit cell determination, and data collection. The goniometer was controlled using the APEX2 software suite, v2008-6.0.51 Integrated intensity information for each reflection was obtained by reduction of the data frames with the program APEX2.51 The absorption correction program SADABS52 was employed to correct the data for absorption effects. A solution was obtained readily using XT/XS in APEX2.51,53,54 Hydrogen atoms were placed in idealized positions and were set riding on the respective parent atoms. All the non-hydrogen atoms were refined with anisotropic thermal parameters. The absence of additional symmetry and voids were confirmed using PLATON (ADDSYM).55 The structure was refined (weighted least-squares refinement on F2) to convergence.53,54,56 Olex2 was employed for the final data presentation and structure plots.333.5 Procedure for enantioselective reduction of ketones
The Ru complexes (1–6) of the type [RuCl2(η6-p-cymene)L] (0.005 mmol) and NaOH (1 mmol) were dissolved in 2-propanol (5 mL). The mixture was stirred at 82 °C for 10 minutes. Then ketone (1 mmol) was introduced into the mixture and continued the stirring at 82 °C. After 10 h, the reaction mixture was cooled to room temperature and then passed through a silica gel short column with n-hexane–ethyl acetate (1:1) eluent to remove the catalyst. The conversions were monitored by GC-MS and GC, and the enantiomeric excesses were calculated by using chiral HPLC.
3.6 Synthesis of (R)-methyl-2-(3-benzoylthioureido)-3-phenylpropanoate
To confirm [RuCl2(η6-p-cymene)]2-catalyzed conversion of amino acid to amino ester, the unprotected D-phenylalanine derived thiourea ligand (L1) (0.5 mmol) and [RuCl2(η6-p-cymene)]2 (0.005 mmol) were dissolved in methanol (10 mL) and the reaction was proceeded at 27 °C. After 12 h, the solvent was evaporated under reduced pressure and the reaction mixture was dissolved in ethyl acetate and passed through a silica gel column to obtain the pure ester. The ester product was confirmed by 1H NMR.O and CS attached N–H), 11.59 (d, 1H, J = 10 Hz, CS attached N–H).4 Conclusions
The new chiral aroylthiourea ligands have been derived from D/L-phenylalanine and reacted with [RuCl2(η6-p-cymene)]2. Ru-p-cymene precursor initially catalyzed the conversion of amino acids to corresponding amino esters, then formed new Ru complexes with the ester of aroylthiourea ligands (1–6). Spectroscopic studies exposed the monodentate sulfur coordination of the aroylthiourea ligand with Ru(II), which was further confirmed by the crystal structure of the ligand L1 and two of the complexes (3 and 6). The chiral Ru-p-cymene complexes (1–6) catalyzed the enantioselective reduction of ketones to their analogous chiral secondary alcohols in the presence of 2-propanol (hydrogen donor) and NaOH (base) with good to excellent conversion and enantioselectivity. The performance of the present catalytic system is superior to most of the existing Ru-p-cymene/amino acid or amino acid derived ligand systems in terms of conversion and ee. Our current focus is on retaining acid group in the Ru-p-cymene complexes to promote the solubility of the complexes and subsequently catalysis in water.Acknowledgements
M. M. S. gratefully acknowledges the Department of Science and Technology, Ministry of Science and Technology, Government of India, for the doctoral fellowship under INSPIRE scheme. We thank Dr S. Velmathi, NIT, Trichy, for polarimetric analysis.References
- G. M. Coppola and H. F. Schuster, Asymmetric Synthesis: Construction of Chiral Molecules Using Amino Acids, Wiley, New York, 1987 Search PubMed.
- N. Selvakumaran, L. Sandhiya, N. S. P. Bhuvanesh, K. Senthilkumar and R. Karvembu, New J. Chem., 2016, 40, 5401–5413 RSC.
- M. Mary Sheeba, S. Preethi, A. Nijamudheen, M. Muthu Tamizh, A. Datta, L. J. Farrugia and R. Karvembu, Catal. Sci. Technol., 2015, 5, 4790–4799 CAS.
- N. Selvakumaran, N. S. P. Bhuvanesh and R. Karvembu, Dalton Trans., 2014, 16395–16410 RSC.
- M. Mary Sheeba, M. Muthu Tamizh, L. J. Farrugia, A. Endo and R. Karvembu, Organometallics, 2014, 33, 540–550 CrossRef.
- N. Gunasekaran, P. Ramesh, M. N. G. Ponnuswamy and R. Karvembu, Dalton Trans., 2011, 12519–12526 RSC.
- K. R. Koch, Coord. Chem. Rev., 2001, 216, 473–488 CrossRef.
- E. Farkas and I. Sovago, Amino Acids, Pept., Proteins, 2014, 39, 21–67 Search PubMed.
- T. G. Appleton, Coord. Chem. Rev., 1997, 166, 313–359 CrossRef CAS.
- A. Iakovidis and N. Hadjiliadis, Coord. Chem. Rev., 1994, 135–136, 17–63 CrossRef.
- K. Ahlford and H. Adolfsson, Catal. Commun., 2011, 12, 1118–1121 CrossRef CAS.
- F. Tinnis and H. Adolfsson, Org. Biomol. Chem., 2010, 8, 4536–4539 CAS.
- J. Wettergren, A. B. Zaitsev and H. Adolfsson, Adv. Synth. Catal., 2007, 349, 2556–2562 CrossRef CAS.
- P. Pelagatti, M. Carcelli, F. Calbiani, C. Cassi, L. Elviri, C. Pelizzi, U. Rizzotti and D. Rogolino, Organometallics, 2005, 24, 5836–5844 CrossRef CAS.
- P. Västilä, J. Wettergren and H. Adolfsson, Chem. Commun., 2005, 2, 4039–4041 RSC.
- I. M. Pastor, P. Västilä and H. Adolfsson, Chem.–Eur. J., 2003, 9, 4031–4045 CrossRef CAS PubMed.
- D. Chatterjee, S. Basak, A. Mitra, A. Sengupta, J. Le Bras and J. Muzart, Inorg. Chim. Acta, 2006, 359, 1325–1328 CrossRef CAS.
- D. Chatterjee, S. Basak, A. Riahi and J. Muzart, J. Mol. Catal. A: Chem., 2006, 255, 283–289 CrossRef CAS.
- J. R. Khusnutdinova, Y. Ben-David and D. Milstein, Angew. Chem., Int. Ed., 2013, 52, 6269–6272 CrossRef CAS PubMed.
- K. Sakthivel, W. Notz, T. Bui and C. F. Barbas, J. Am. Chem. Soc., 2001, 123, 5260–5267 CrossRef CAS PubMed.
- Y. Zhao, J. Rodrigo, A. H. Hoveyda and M. L. Snapper, Nature, 2006, 443, 67–70 CrossRef CAS PubMed.
- B. M. Trost and K. Dogra, J. Am. Chem. Soc., 2002, 124, 7256–7257 CrossRef CAS PubMed.
- J. W. Faller and A. R. Lavoie, Organometallics, 2001, 20, 5245–5247 CrossRef CAS.
- A. B. Zaitsev and H. Adolfsson, Org. Lett., 2006, 8, 5129–5132 CrossRef CAS PubMed.
- T. Ohta, S. Nakahara, Y. Shigemura, K. Hattori and I. Furukawa, Appl. Organomet. Chem., 2001, 15, 699–709 CrossRef CAS.
- X.-N. Li, H.-Y. Zhou, L. Feng, K. Duan and J.-X. Wang, Appl. Organomet. Chem., 2012, 26, 168–174 CrossRef CAS.
- A. Bøgevig, I. M. Pastor and H. Adolfsson, Chem.–Eur. J., 2004, 10, 294–302 CrossRef PubMed.
- P. Kumar, R. K. Gupta and D. S. Pandey, Chem. Soc. Rev., 2014, 43, 707–733 RSC.
- H. Cao, L.-H. Cai, C.-X. Wang, X.-H. Zhu, Z.-M. Li and X.-F. J. Hou, J. Organomet. Chem., 2015, 775, 60–66 CrossRef CAS.
- S. K. Manna, S. L. K. Manda and G. Panda, Tetrahedron Lett., 2014, 55, 5759–5763 CrossRef CAS.
- B. S. Vig, P. J. Lorenzi, S. Mittal, C. P. Landowski, H.-C. Shin, H. I. Mosberg, J. M. Hilfinger and G. L. Amidon, Pharm. Res., 2003, 20, 1381–1388 CrossRef CAS.
- G. C. Barrett, Amino Acids, Peptides and Proteins, RSC Publications, London, 1996, vol. 27, p. 1 Search PubMed.
- N. Atsushi, M. Toyoharu, K. Hiroto and E. Takeshi, Macromolecules, 2003, 36, 9335–9339 CrossRef.
- S. Fumio and E. Takeshi, Macromol. Chem. Phys., 1999, 200, 2651–2661 CrossRef.
- J. Kim, J. Kim, S. Song, O. Jung and H. Suh, J. Inclusion Phenom. Macrocyclic Chem., 2007, 58, 187–192 CrossRef CAS.
- G. Pollini, N. Baricordi, S. Benetti, C. De Risi and V. Zanirato, Tetrahedron Lett., 2005, 46, 3699–3701 CrossRef CAS.
- C. Somlai, A. Peter, P. Forgo and B. Penke, Synth. Commun., 2003, 33, 1815–1820 CrossRef CAS.
- Z. Tomasz, A. Michał and J. Janusz, Tetrahedron: Asymmetry, 2002, 13, 2053–2059 CrossRef.
- V. K. Tandon, D. B. Yadav, R. V. Singh, A. K. Chaturvedi and P. K. Shukla, Bioorg. Med. Chem. Lett., 2005, 15, 5324–5328 CrossRef CAS PubMed.
- S. L. Manjinder, K. R. Yeeman, N. G. J. Michael and C. V. John, J. Org. Chem., 2002, 67, 1536–1547 CrossRef.
- E. Pękala and D. Żelaszczyk, Sci. Pharm., 2009, 77, 9–17 CrossRef.
- S. T. Marino, D. Stachurska-Buczek, D. A. Huggins, B. M. Krywult, C. S. Sheehan, T. Nguyen, N. Choi, J. G. Parsons, P. G. Griffiths, I. W. James, A. M. Bray, J. M. White and R. S. Boyce, Molecules, 2004, 9, 405–426 CrossRef CAS PubMed.
- R. Noyori and T. Ohkuma, Angew. Chem., Int. Ed., 2001, 40, 40–73 CrossRef CAS.
- Chirality in Industry: The Commercial Manufacture and Applications of Optically Active Compounds, ed. A. N. Collins, G. N. Sheldrake and J. Crosby, John Wiley & Sons, Chichester, 1997 Search PubMed.
- A. Barth, Prog. Biophys. Mol. Biol., 2000, 74, 141–173 CrossRef CAS PubMed.
- X.-J. Zhang, T.-B. Lai and R. Y.-C. Kong, Top. Curr. Chem., 2011, 308, 365–404 CrossRef.
- S. D. Taylor, C. C. Kotoris and G. Hum, Tetrahedron, 1999, 55, 12431–12477 CrossRef CAS.
- T. J. Barbarich, C. D. Rithner, S. M. Miller, O. P. Anderson and S. H. Strauss, J. Am. Chem. Soc., 1999, 121, 4280–4281 CrossRef CAS.
- J. Wu, J.-X. Ji and A. S. C. Chan, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 3570–3575 CrossRef CAS PubMed.
- M. A. Bennet, T. N. Huang, T. W. Matheson and A. K. Smith, Inorg. Synth., 1982, 21, 74–78 CrossRef.
- APEX2 and APEX3 Program for Data Collection on Area Detectors, BRUKER AXS Inc., 5465 East Cheryl Parkway, Madison, WI 53711-5373, USA Search PubMed.
- G. M. Sheldrick, SADABS, Program for Absorption Correction of Area Detector Frames, BRUKER AXS Inc., 5465 East Cheryl Parkway, Madison, WI 53711-5373, USA Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- A. L. J. Spek, Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: A table of the X-ray crystallographic data, atomic coordinates in CIF format, 1H NMR and 13C NMR spectra of all the ligands and complexes, and GC-MS, GC and HPLC data are included. CCDC 1474599, 1458027–1458028. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra12428c |
| This journal is © The Royal Society of Chemistry 2016 |