
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A computational study of the interaction of organic surfactants with goethite α-FeO(OH) surfaces†
David Santos-Carballal *ab,
Zhimei Dub,
Helen E. Kingc and
Nora H. de Leeuw*abc
*ab,
Zhimei Dub,
Helen E. Kingc and
Nora H. de Leeuw*abc
aSchool of Chemistry, Cardiff University, Main Building, Park Place, Cardiff CF10 3AT, UK. E-mail: SantosCarballalD@cardiff.ac.uk; DeLeeuwN@cardiff.ac.uk; Tel: +44 (0)29 2087 0827 Tel: +44 (0)29 2087 0658
bDepartment of Chemistry, University College London, 20 Gordon Street, London WC1H 0AJ, UK
cDepartment of Earth Sciences, Utrecht University, Budapestlaan 4, 3584 CD Utrecht, The Netherlands
First published on 9th September 2016
Abstract
We have studied the adsorption of three organic molecules onto different surfaces of goethite α-FeO(OH) using atomistic simulation techniques. New interatomic potentials for the interaction between goethite and the organic molecules were developed. In the majority of cases the organic molecules were found to be capable of forming a coordinate bond via their carbonyl oxygen atom with a surface iron ion. In addition, weaker hydrogen bonds were formed between the organic molecules and the surfaces. The largest adsorption energies were obtained for the modes of adsorption where the organic molecules bridged or spanned the periodic grooves or dips present on the goethite surfaces, thus forming several interactions between the molecule and the surface. Among all adsorbates studied, the hydroxamic acid molecule in the eclipsed conformation releases the largest adsorption energy when it interacts with goethite surfaces, followed by the staggered conformations of hydroxyethanal and methanoic acid molecules. The adsorption energies are in the range of −60.0 to −186.4 kJ mol−1. Due to the surface structure, as well as the flexibility and size of hydroxamic acid and hydroxyethanal, in most cases these adsorbate molecules lose their planarity with respect to the structure of the isolated molecules. We found that the replacement of pre-adsorbed water by the organic adsorbates is an exothermic process on all the goethite surfaces studied. The removal by sorption onto iron particles of humic and fulvic acids, the major substituents of natural organic matter (NOM) that pollutes aquifers and soils, is corroborated by our calculations of the adsorption of surfactants with the same functional groups as the surfaces of oxidised iron particles.
1. Introduction
After silicon and aluminium, iron is the third most abundant cation in the crust of the Earth. It is found mainly in soils, sediments and rocks as (hydr)oxides, such as goethite (α-FeO(OH)) and hematite (α-Fe2O3), which are the most common forms found in cool-temperate and warm climates respectively.1 These iron (hydr)oxides have also been found coating other naturally occurring minerals such as quartz and clay2 and they are also the most common product of iron metal corrosion in aqueous environments,3 as well as in various applications that use zero-valent iron (ZVI) as a reductant e.g., in the treatment of soils4 and aquifers.5 In environmental remediation, iron (hydr)oxides themselves have been reported to act as powerful removers of many contaminants, including heavy metals6–8 and organic matter.9,10Although pollutants have been traditionally disposed of in landfill sites, the disastrous consequences for humans from leaking chemical waste, as for example observed in Love Canal in the USA and Lekkerkerk in Holland,11 have forced governments to find methods of transforming pollutants into less harmful forms to reduce the hazards associated with their disposal.12 Therefore, the goal of remediation processes is to destroy or degrade the pollutant; extract it or isolate it from non-contaminated material to allow it to be treated afterwards; stabilise it in a less toxic or mobile form; or contain it to avoid damage to the environment.13 Often the remediation of both soil and groundwater must be accomplished simultaneously due to their constant interaction.14 To this end, biological,15–17 chemical18–20 and physical21–23 approaches are employed, usually in combination to achieve the best results.24 A good example of a low-maintenance and -cost remediation technology is the ‘Iron wall’, where ZVI particles are applied. These ‘walls’ are composed of approximately 80% sand and 20% granular iron, where the latter is responsible for pollutant removal.5 The pathway for pollutant removal from aqueous solutions by ZVI particles can be represented as: (1) oxidation of the iron particle surfaces, (2) adsorption of the pollutant to the iron oxide surface, (3) formation of a complex between the pollutant and the surface iron ions and (4) reduction of the contaminant at the expense of the ZVI particle.10 This series of events is supported by experimental evidence of the core–shell structure found in ZVI particles, where the shell is composed of iron (hydr)oxides and the core of metallic iron that is slowly oxidised to supply electrons for the reduction of adsorbed pollutants on the shell.8 As one of the primary steps in the synergic remediation mechanism of adsorption–reduction operating in the ‘Iron wall’ technology, the selective adsorption of the pollutants to the iron (hydr)oxides in the shell of the ZVI particles determines the effectiveness of the contaminant removal and decomposition.
A number of previous studies have described the interaction between iron (hydr)oxide and various types of molecules such as water,25 inorganic ligands,26 organic acids,27–37 siderophore azotobactin,38 and natural organic matter (NOM).39–43 Despite the added effects of the pH of the medium, the adsorbate molecular mass,41,42 competing ionic sorbates and ionic strength, the adsorption to iron oxides is determined mainly by the presence and type of functional groups in the adsorbate.42 Carboxyl and the phenol hydroxyl groups present in NOM are the main functional groups responsible for the adsorption onto metal oxides.28,39,41,44,45 The most polar moieties of aliphatic molecules are adsorbed onto the iron oxide surfaces by electrostatic attractions or ligand exchange reactions.43 The aim of this work, therefore, is to carry out atomistic simulations to study the adsorption modes and strength of interaction of three organic molecules with representative functional groups with major goethite surfaces, where one hydroxylated aldehyde and two organic acids are selected as models for organic pollutants, i.e. methanoic acid, hydroxyethanal and hydroxamic acid. This selection of adsorbates will give us the opportunity to determine the effect of the chain length, different functional groups as well as adsorbate conformation on the adsorption process.46 In addition, these organic acid molecules are simple models for the carboxylic type of ligands present in humic and fulvic acids,45 which are the main substituents of NOM, one of the major contaminants present in aquifers and soils.41 The structural differences in the vicinity of the carbonylic group in these organic acid molecules will allow us to investigate the heterogeneity of the acids present in NOM.
2. Methodology
Interatomic potential-based simulations were performed using the METADISE code,47 to model the structures and calculate the energies of the goethite surfaces, adsorbate molecules and their adsorption. These energy minimisation techniques are based on the Born model of solids, which treats the interactions between the ions in the crystal as long-range electrostatic and short-range forces.48 According to this model, the former type of interactions are simply the Coulomb sum of electrostatic forces calculated using the Parry technique,49,50 which is an excellent method for describing systems with surfaces, while the latter are described by parameterised analytical equations that include repulsion and van der Waals attractions between neighbouring electron charge clouds. All geometry optimizations were carried out using static minimisation methodologies. The conjugate gradients method was used in the initial iterations of the minimisation to roughly guide the system to the zone where the energy has a harmonic dependence on the position of the ions,51 followed by the Newton–Raphson method which is more suitable near the energy minimum.52 The energy minimisation techniques adjusted the atomic positions until the net forces on each atom were zero.47 The electronic polarisability of the ions, in our case the different oxygen species in goethite, is represented by the shell model of Dick and Overhauser, which considers the polarisable ion as a massy core with a massless shell connected by a harmonic spring.53 This model considers that the polarisability of an ion is determined by the relative charges between core and shell and the constant of the spring that joins them.2.1. Interatomic potential model
Three sets of potential parameters have been used in this work, which are fully compatible. The parameters developed by de Leeuw and Cooper have been used for modelling the goethite (α-FeO(OH)).25 The interatomic potentials for goethite are based on the Fe(III) oxide parameters of Lewis and Catlow54 with the polarisable hydroxy potential of Baram and Parker55 and the short-range interactions between the iron ion and the hydroxy oxygen scaled according to Schröder et al.56 A selection of parameters from the consistent valence force field (CVFF) was chosen for the organic molecules,57 which have been used in a number of previous studies.46,58,59 The interaction parameters for the organic molecules with the goethite surfaces were derived following the method of Schröder et al.,56 where the short-range Buckingham potentials were scaled according to the electronic charges on the atoms of the organic molecule. The detailed interaction parameters used in this work are listed in Tables ESI-1, ESI-2 and ESI-3 of the ESI.†Previous studies60–64 have shown good agreement between the adsorption energies calculated using energy minimisation simulations and experimental techniques such as adsorption calorimetry,65 as well as identification of the configurations with largest binding energies for the interaction of water with CaO and silica. Moreover, earlier predictions, using atomistic simulations, of morphologies as well as hydration energies of several calcium carbonates,66 were later confirmed by experimental techniques.67 For the adsorption structures, interatomic potential calculations usually provide good results, as shown by atomistic simulation studies in combination with ab initio methods such as density functional theory (DFT). Previous studies of several different materials, such as α-quartz,64 calcium fluoride,68 hydroxyapatite69–72 and scheelite,73 have indicated that there is good agreement between DFT calculations and interatomic potential-based simulations of modes and energies of the adsorption of small to medium-sized molecules. We are therefore confident that the interatomic potentials derived here for the interaction of goethite with the organic molecules are appropriate for the calculation of adsorption energies and structures.
2.2. Adsorption energies
The adsorption of isolated organic molecules onto each surface was modelled, but to ensure that a large number of interfacial configurations were tested, a comprehensive range of initial positions of the organic molecules with respect to each surface was sampled, including a series of heights, orientations and lateral displacements. The vertical distance between the organic molecule and the surface varied from 1.5 Å to 3.5 Å in 1.0 Å intervals. For each height, 72 orientations of the molecule towards the surface were inspected, which included 12 rotations of 30° in the axis perpendicular to the surface for the molecule lying parallel or perpendicular to the surface. Prior to the energy minimisation, a systematic scanning procedure was carried out, where the organic molecule was kept at a constant height above the surface and moved in two directions over the surface without relaxing the surface or the molecule. The energy for the unrelaxed system was calculated as a series of points on a grid. This grid was determined by the surface lattice vectors at intervals of 0.5 Å. This scanning process supplied information on the interfacial energy at each point on the grid, hence identifying the lateral displacement with largest binding energy of the centre of mass of the molecule with respect to the surface for the unrelaxed interfacial system. The 10 points of the grid with the largest binding energies from each scan were then used as the starting configurations for a full geometry optimisation.In order to model the adsorption, the simulation cells were constructed so that the surfaces were large enough to accommodate the organic molecules without any constraints to the geometry of the organic molecule or interference between the organic molecule and its images in the periodically repeated surface cell.
The adsorption energies (Eads) were calculated by using eqn (1).
| Eads = Esystem − (Esurf + Eadsorbate) | (1) |
3. Results and discussion
3.1. Goethite
Goethite (α-FeO(OH)) has a typical orthorhombic structure with space group Pbnm. The goethite structure is shown in Fig. 1, where the oxygen ions are in hexagonal close packed arrangement along the [100] direction and the Fe(III) ions occupy half of the octahedral sites between two layers. Consequently, the Fe(III) ions are stacked as double rows separated by double rows of empty sites, which appear as grooves in the surface or tunnels in the bulk.1 In addition, goethite has tetrahedral interstices between three oxygen or hydroxy oxygen ions in one plane and those in the plane above. The goethite structure determined by Hazemann with α = β = γ = 90° and a = 4.616 Å, b = 9.955 Å and c = 3.023 Å was used as our starting structure.74 The lattice parameters of the relaxed goethite bulk crystal were α = β = γ = 90°, a = 4.677 Å, b = 9.583 Å and c = 3.111 Å.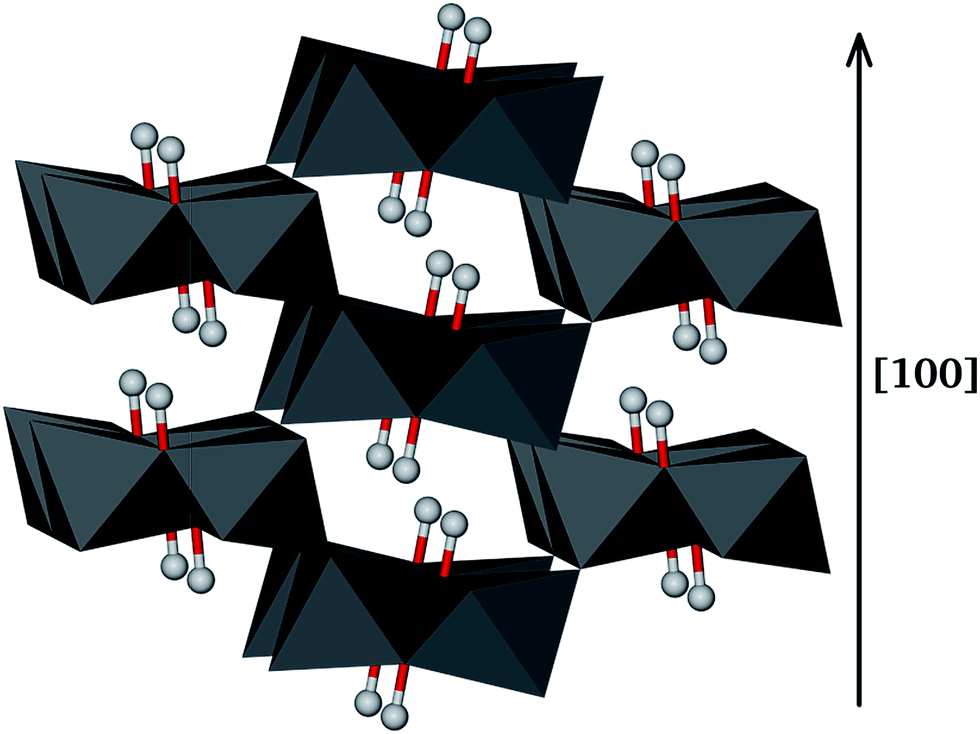 | ||
| Fig. 1 Arrangement of double chains formed by edge-sharing octahedra in the [100] direction, where each iron ion is surrounded by three O2− and three OH− ions. Hydrogen atoms are also shown as balls. | ||
The goethite morphology has been the subject of both previous experimental75 and computational25 studies. The most prominent mineral surfaces reported are {010} and {101}, where the {010} surface is the natural cleavage plane of goethite. The full coordination of the oxygen atoms on the {010} surface also makes it the most stable goethite surface.25 The {001}, {100}, {011} and {111} surfaces have also been included in this study. These surfaces are expected in the natural environment where goethite crystals can be found,1 although they are less stable than the {010} or {101} as the coordination number of the surface oxygen is two, compared to a bulk coordination of three, and the iron coordination number is decreased from six to four.25 The selection of surfaces provides a number of different surface structures to investigate the strength and modes of interaction with the model pollutants. In accordance with previous studies, the hydroxy group of goethite is considered as an anion with covalent character.25 Therefore, the hydrogen and oxygen atoms of the hydroxy groups have been kept together when creating the surfaces. The α-FeO(OH){001}, {010}, {011}, {100}, {110} and {111} surfaces were modelled using cells with surface areas of 448.21, 509.07, 471.22, 417.26, 464.29 and 446.05 Å2, respectively.
3.2. Organic adsorbates
Determination of the key aspects in the competition of pollutants for binding sites on adsorbents plays an important role in modelling their interaction.76 From previous studies, it is known that the strength of adsorption of molecules with common carboxylic acid and amide organic functional groups is highly dependent on their structures. However, the exact behaviour of each adsorbent/adsorbate pair is not known.39 In this work, adsorption of three different organic molecules to the goethite surfaces was studied, namely, methanoic acid (HCOOH), hydroxyethanal (HC(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)CH2OH) and hydroxamic acid (HC(O)NHOH) (see Fig. 2). The methanoic acid and hydroxyethanal molecules provide the opportunity to study the effect of chain length and separation of the CO and OH groups on adsorption. The hydroxamic acid molecule enables us to study the influence of the replacement of a carbon by a nitrogen atom on their affinity for the goethite surfaces. Two extreme conformers have been considered for both hydroxyethanal and hydroxamic acid, one with the oxygen atoms as far away as possible from each other, which was labelled as staggered hydroxyethanal and hydroxamic acid, and the other with the two oxygen atoms eclipsed, called eclipsed hydroxyethanal and hydroxamic acid. There is evidence that hydroxamic acid and hydroxyethanal rotate through the C–N and C–C bond, respectively, during minimization in order to accommodate the carbonyl oxygen atom and the hydroxy group facing the adsorbant surface to maximize the number of interactions between them.46,58,77,78
O)CH2OH) and hydroxamic acid (HC(O)NHOH) (see Fig. 2). The methanoic acid and hydroxyethanal molecules provide the opportunity to study the effect of chain length and separation of the CO and OH groups on adsorption. The hydroxamic acid molecule enables us to study the influence of the replacement of a carbon by a nitrogen atom on their affinity for the goethite surfaces. Two extreme conformers have been considered for both hydroxyethanal and hydroxamic acid, one with the oxygen atoms as far away as possible from each other, which was labelled as staggered hydroxyethanal and hydroxamic acid, and the other with the two oxygen atoms eclipsed, called eclipsed hydroxyethanal and hydroxamic acid. There is evidence that hydroxamic acid and hydroxyethanal rotate through the C–N and C–C bond, respectively, during minimization in order to accommodate the carbonyl oxygen atom and the hydroxy group facing the adsorbant surface to maximize the number of interactions between them.46,58,77,78
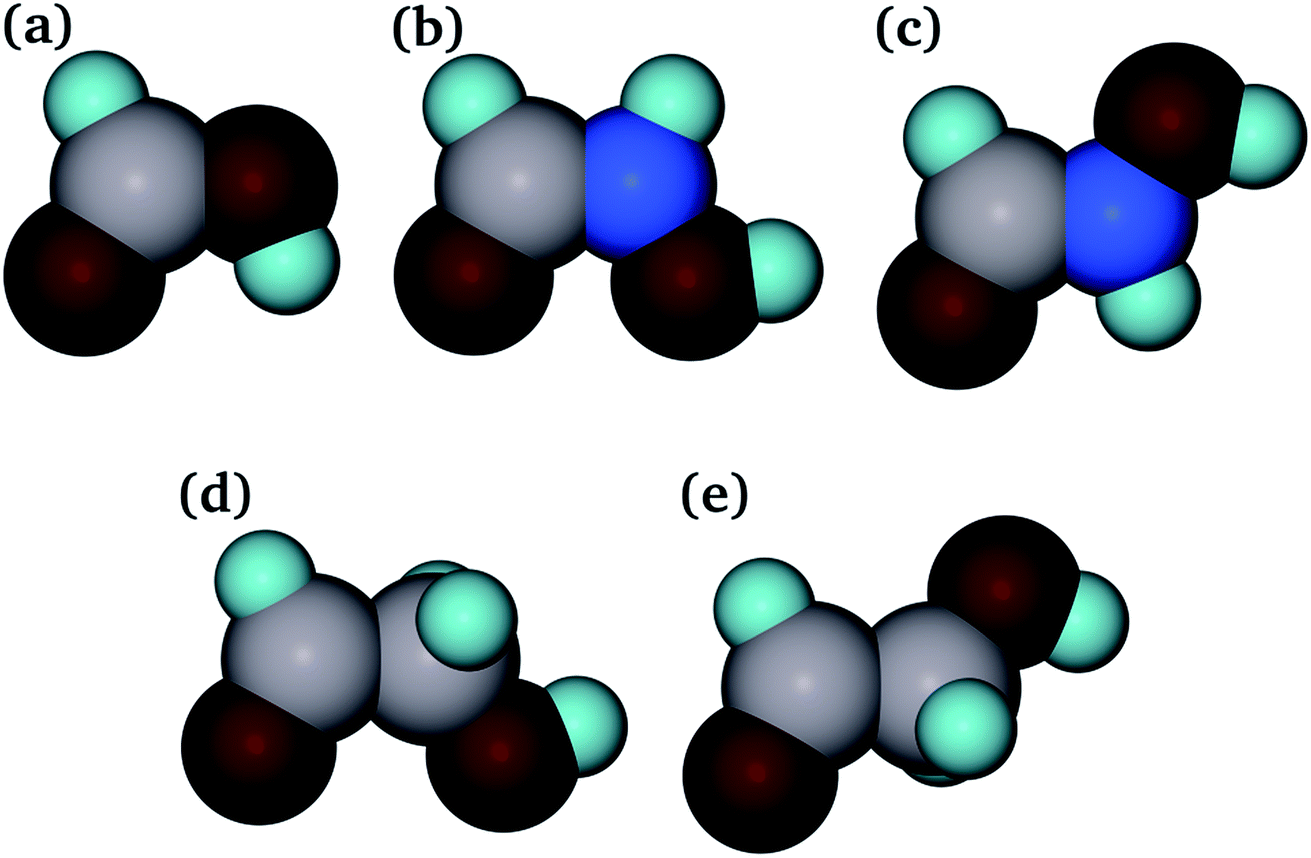 | ||
| Fig. 2 (a) Methanoic acid, (b) eclipsed hydroxamic acid, (c) staggered hydroxamic acid, (d) eclipsed hydroxyethanal and (e) staggered hydroxyethanal (O, dark red; H, pale blue; C, pale grey; N, bright blue). | ||
Additionally, the hydroxy hydrogen atom rotated freely during geometry optimisation of the organic molecules, either when they were isolated or when interacting with the surface. In order to search for the structure with the largest interfacial energy and avoid finding a local minimum, the goethite surfaces have been scanned with every adsorbate in their different conformations with various orientations and heights towards the surfaces. Based on previous studies, which have shown that the use of non-dissociated molecules in the calculation of adsorption trends is in line with the experimental behaviour,79 the dissociation of the organic molecules was not considered in this study.
3.3. Scan
For each interfacial system, the organic adsorbate was scanned at 3 different heights and 72 orientations with respect to the goethite surface at each height. Given the size of the surface simulations cells, the adsorbate molecule was never closer than 19.17 Å or farther than 23.38 Å of its periodically repeated image, which ensured that no interactions occurred between them. Here, the system of the goethite {001} surface with the staggered conformation of the hydroxamic acid molecule is used to explain the scanning procedure. For this pair, the contour plot presented in Fig. 3 indicates that the unrelaxed adsorption energy is highly dependent on their relative positions. Fig. 3(a) and (b) show the unrelaxed adsorption energy map for the molecule at 2.5 Å above the surface, where valleys and hills are shifted and their topological features are changed as a result of the variation of the relative surface/adsorbate orientation, although their energy scales are essentially the same. Compared to a 0° orientation, for a molecule with 60° rotation the regions with the largest unrelaxed adsorption energy move from the surface Fe atoms (see Fig. 3(a)) to areas between the oxygen atoms in three neighbouring FeO(OH) units (see Fig. 3(b)). The results of the scan at two different heights (see Fig. 3(c) and (d)) show that the height has more impact than orientation on the unrelaxed adsorption energy. When the centre of mass of the molecule is kept at 1.5 Å from the surface, the energy scale becomes 430 times larger and the most repulsive regions are located between the oxygen atoms in three FeO(OH) neighbour units (see Fig. 3(c)). However, when the mass centre of the hydroxamic acid molecule is 3.5 Å above the surface, the regions between the same three oxygen atoms are transformed into the areas with the largest binding energies of the unrelaxed system, reducing the energy scale significantly (see Fig. 3(d)). The change in the energy scale in the contour maps with the distance between the surfaces and the adsorbate can be rationalized in terms of the typical interatomic distances between the organic molecule and the surface atoms found for the interaction in this study, which are discussed in detail in subsequent sections. 1.5 Å and 3.5 Å are shorter or longer respectively than typical interatomic distances for the interaction between the molecules and surfaces, resulting in high repulsive or low positive unrelaxed adsorption energies. The optimal distance between the centre of mass of the molecule and the surface is 2.5 Å, where the energy scale has small negative to positive values.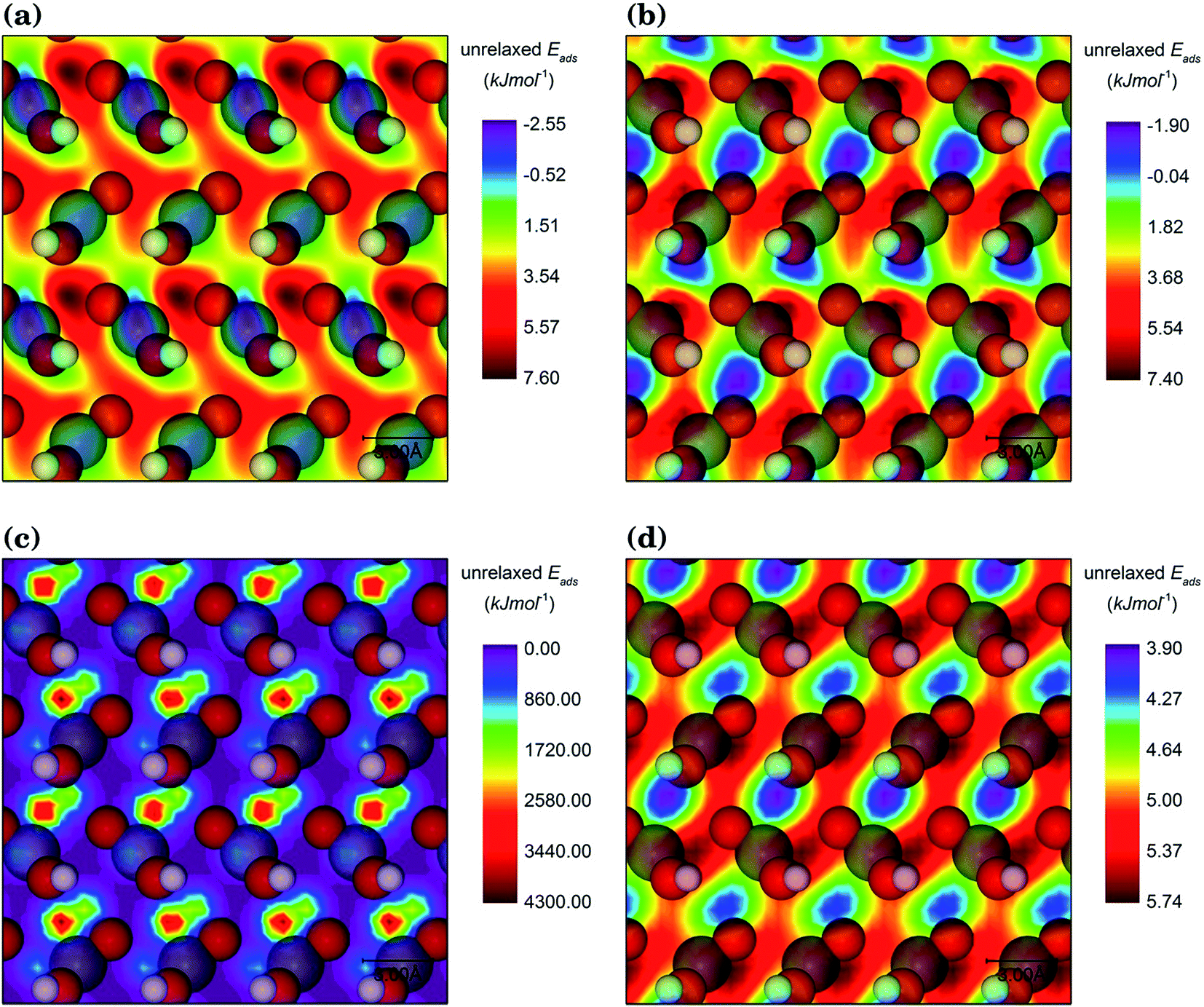 | ||
| Fig. 3 Contour plot of the interfacial energies of the unrelaxed systems of the staggered conformation of hydroxamic acid molecule onto the goethite {001} surface with (a) 0° and (b) 60° of rotation of the molecule at 2.5 Å distance and (c) at 1.5 and (d) 3.5 Å distances in a fixed orientation. Surface atoms are also shown (O, red; H, white; Fe, grey). | ||
Similar calculations were conducted for other goethite/organic adsorbates systems in order to corroborate the applicability of the preferred distance for all orientations. For each interfacial system, the 10 scanned positions of the molecule with the largest negative unrelaxed adsorption energies were used as the starting configuration for the full geometry optimisations described in Section 3.4.
3.4. Adsorption structures
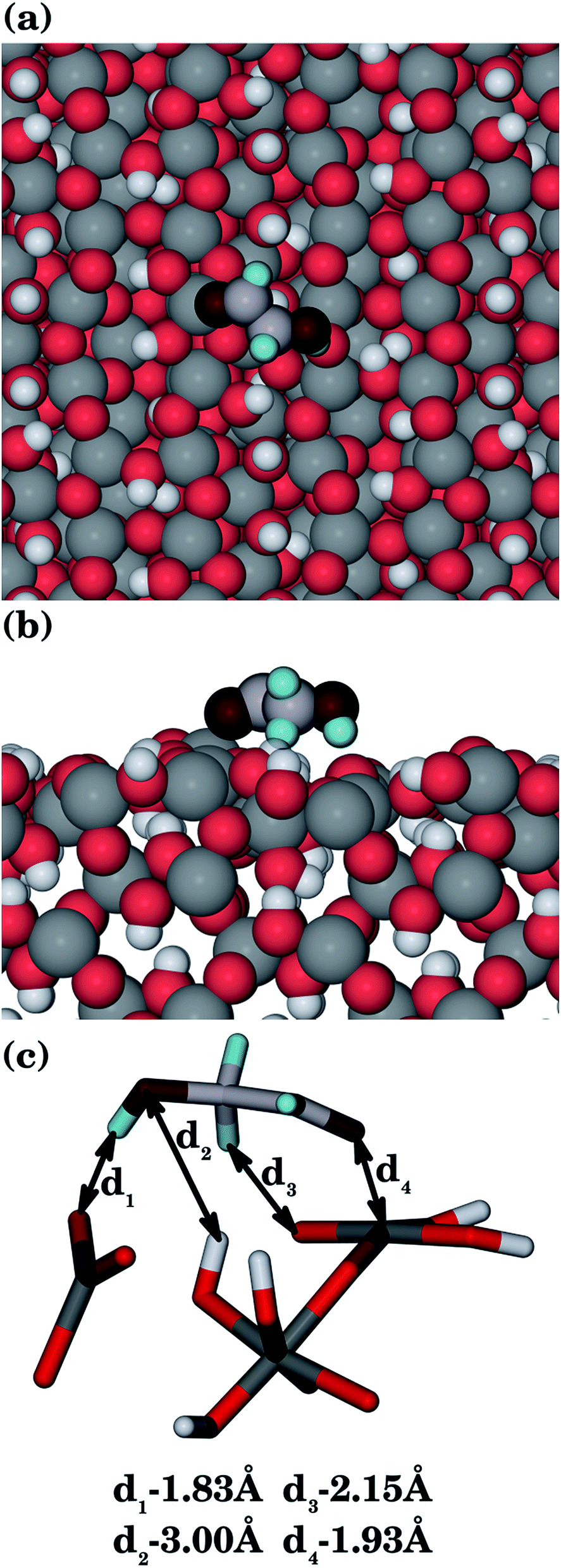
The most important features of the adsorption modes, namely the interatomic distances between the organic molecules and all goethite surfaces are given in Table 1. Our results indicate that there is electrostatic character in several of the most favourable interactions between the surfaces and the organic molecules, which allows the main centres of adsorption on the surfaces (usually the iron ions) to interact with their counterparts in the organic molecules (oxygen and nitrogen atoms). The interactions are not only given by the coordination between atoms of opposite charge, but also for the hydrogen-bonds between the surface and the organic molecules. The adsorption energies mainly depend on the coordination between the negative atoms of the organic molecule and the iron ions of the surfaces. However, the number and strength of the hydrogen-bonds will make an important secondary contribution to the final adsorption energy if the coordination to the iron ions is similar.| Surfactant | Surface | Fe–O |
Fe–OH | Fe–N | Osurf–H (O, C, N) | OHsurf–H (O, C, N) | Hsurf–(O, N) |
|---|---|---|---|---|---|---|---|
| HCOOH | {010} | 2.08 | 1.95 | 2.56 | |||
| {110} | 2.04 | 2.02 | 2.64, 2.82 | 2.45, 2.49, 2.68 | |||
| {111} | 2.31, 2.49 | 1.75, 2.44, 2.95 | 2.70, 2.76, 2.85, 2.97 | ||||
| {001} | 1.87 | 2.88 | 2.08, 2.51 | ||||
| {011} | 2.60, 2.88 | 2.06 | 2.02, 2.86 | 2.48, 2.89, 2.99 | |||
| {100} | 2.01 | 2.70 | 2.64 | 1.95, 2.88 | |||
| HC(O)NHOH |
{010} | 2.15 | 2.45 | 1.97 | 2.07 | 2.56, 2.68 | |
| {110} | 1.97 | 1.89, 2.24, 2.99 | 2.32, 2.39 | 2.72, 2.83 | |||
| {111} | 2.00 | 2.47, 2.94 | 2.81, 2.89 | 1.74, 2.17, 2.98 | 2.78, 2.96 | 2.78, 2.86, 2.87 | |
| {001} | 1.82 | 2.36 | 2.75 | 2.89 | 1.97, 2.38 | 2.57 | |
| {011} | 1.94, 2.98 | 2.80 | 2.60 | 1.90 | 1.97, 2.09, 2.86, 2.90 | 2.84, 2.85, 2.87 | |
| {100} | 1.98 | 2.91 | 2.05, 2.33 | 2.26, 2.56, 2.73 | 2.49 | ||
| HC(O)CH2OH |
{010} | 2.02 | 2.29 | 2.16, 2.36, 2.76, 2.86, 2.91 | 2.68, 2.57, 2.85, 2.59 | ||
| {110} | 1.97 | 2.11 | 2.40 | 2.48 | 2.49, 2.57, 2.77, 2.89, 2.98 | ||
| {111} | 1.95 | 2.74, 2.88 | 2.22, 2.33 | 2.49, 2.70 × 2, 2.79 | 2.57 | ||
| {001} | 1.83 | 2.11, 2.18 | 2.23, 2.27, 2.84 | 2.94 | |||
| {011} | 2.07 | 2.48 | 1.81 | 2.58, 2.64 | 2.48, 2.70, 2.86 | ||
| {100} | 1.93 | 1.83, 2.15 | 3.00 |
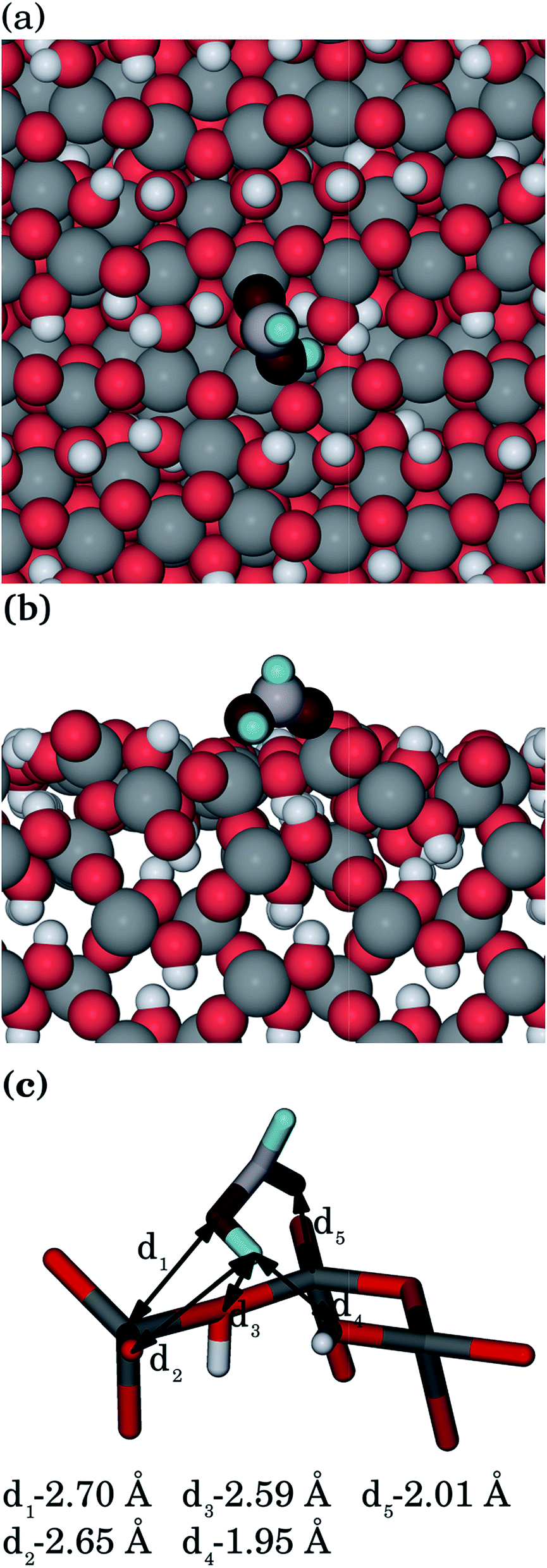 | ||
| Fig. 4 Methanoic acid adsorbed to the goethite {100} surface, showing: (a) top view, (b) side view and (c) interatomic distances (Ogoethite, red; Omethanoic acid, dark red; Hgoethite, white; Hmethanoic acid, pale blue; C, pale grey; Fe, grey). | ||
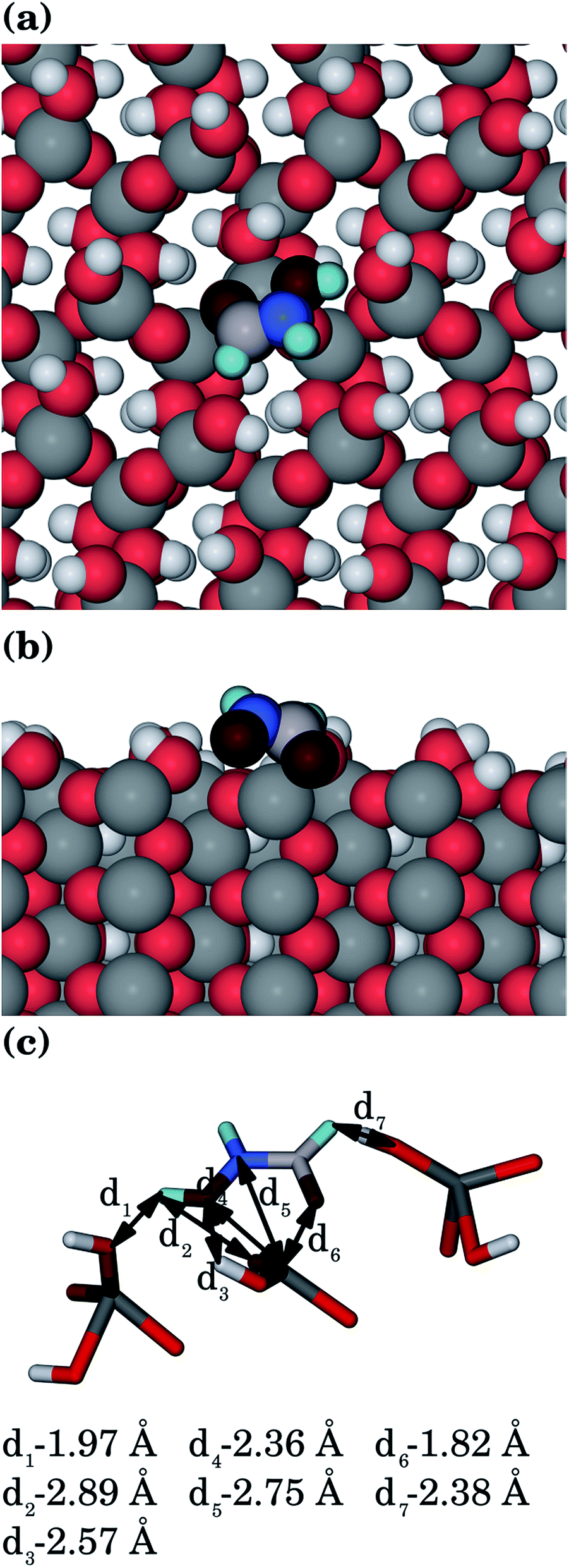 | ||
| Fig. 5 Hydroxamic acid adsorbed to the goethite {001} surface, showing: (a) top view, (b) side view and (c) interatomic distances (Ogoethite, red; Ohydroxamic acid, dark red; Hgoethite, white; Hhydroxamic acid, pale blue; C, pale grey; Fe, grey; N, blue). | ||
| Goethite | Methanoic acid (kJ mol−1) | Hydroxamic acid (kJ mol−1) | Hydroxyethanal (kJ mol−1) | ||||
|---|---|---|---|---|---|---|---|
| Miller index | Surface energya (J m−2) | Hydration energya (kJ mol−1) | Eclipsed | Staggered | Eclipsed | Staggered | |
| a Ref. 25. | |||||||
| {010} | 0.68 | −25.9 | −60.00 | −76.08 | −71.20 | −80.90 | −80.90 |
| {110} | 1.18 | −34.6 | −101.40 | −108.67 | −125.63 | −129.30 | −124.80 |
| {111} | 1.33 | −43.7 | −101.00 | −140.53 | −132.79 | −128.90 | −128.90 |
| {001} | 1.68 | −80.2 | −111.40 | −186.38 | −184.90 | −132.90 | −132.90 |
| {011} | 1.72 | −58.4 | −97.10 | −130.01 | −135.64 | −103.40 | −92.17 |
| {100} | 1.92 | −56.5 | −122.80 | −175.13 | −175.84 | −145.70 | −145.70 |
 | ||
| Fig. 6 Hydroxyethanal adsorbed to the goethite {100} surface, showing: (a) top view, (b) side view and (c) interatomic distances (Ogoethite, red; Ohydroxyethanal, dark red; Hgoethite, white; Hhydroxyethanal, pale blue; C, pale grey; Fe, grey). | ||
3.5. Adsorption energies
Table 2 lists the calculated adsorption energies of the three organic molecules on the goethite surfaces with low Miller indices along with the surface energies, showing a correlation between the surface and the adsorption energies of the organic molecules. It is clear that the {100} and {001}, which are among the less stable surfaces, are the most favourable surfaces for adsorption of all surfactant molecules. On both these surfaces, the protruding hydroxy groups and under-coordinated iron ions contribute to the surface reactivity and lead to relatively larger adsorption energies than the other goethite surfaces studied here. These under-coordinated iron ions produce favourable sites for the carbonyl oxygen atom to bind to with the protruding hydroxy groups taking positions that resemble the arrangement of equivalent hydroxy groups within the goethite bulk, highlighting the importance of the surface structure on the adsorption of the surfactant molecules.The methanoic acid is the least favourable adsorbate in terms of its strength of interaction with the goethite surfaces, which is mainly due to its single functional group that is able to interact with the surfaces. Although it has a carbonylic group, which interacts with the surface species via its oxygen as observed with the other surfactant molecules, the atoms of its hydroxy group have less flexibility due to the direct link of this group to the carbonyl carbon atom. This results in the methanoic acid molecule forming fewer additional weaker interactions with the surface when bridging between different surface species.
The adsorption strength of hydroxy amide (HNOH) or carbinol (H2COH) functional groups can be evaluated by comparing the adsorption of the hydroxamic acid and hydroxyethanal adsorbates, both of which have the HNOH or H2COH group directly linked to the carbonyl group (CO). Considering the carbonyl group as essentially the same for the two adsorbates, the differences in strength and modes of adsorption between these two molecules can be modelled due to the different types of atom linking the carbonyl carbon and the hydroxy oxygen atom (N and C in hydroxamic acid and hydroxyethanal respectively). As mentioned earlier, relatively larger adsorption energies are released when these two surfactants adsorb onto goethite surfaces. In most cases, the adsorption energies of hydroxamic acid are slightly larger than those of hydroxyethanal. The absence of a hydrogen and the presence of nitrogen instead of one of the carbon atoms in the hydroxamic acid enables this molecule to form a number of additional interactions with the surfaces. For example, hydrogen-bonds can form between the hydroxy group species in the organic molecule and the surfaces, enhancing the adsorption of the hydroxide amide with respect to the carbonyl group.
The importance of the proximity of hydroxy and carbonyl group in the surfactant molecules for the strength of adsorption can be assessed by comparing the adsorption energies of methanoic acid and hydroxyethanal onto the goethite surfaces. Table 2 indicates that adsorption of hydroxyethanal on goethite surfaces is always stronger than that of methanoic acid. The differences in the energies can be explained by the capacity of each surfactant to form additional interactions via the hydroxy group atoms. The greater flexibility of the hydroxy group in hydroxyethanal allows it to form stronger interactions with surface species. However, in methanoic acid the hydroxy group is directly attached to the carbonyl carbon making it difficult for this group to interact with the surface.
3.6. Competitive adsorption
Although the remediation process of aquifers and soils occurs under aqueous conditions, thus far the surfaces studied have been anhydrous. As remediation processes take place in an aqueous environment, it is important not only to understand the adsorption strength of these organic molecules on the dry surfaces of goethite, but also on the hydrated surfaces. The remediation process can be seen as the displacement of pre-adsorbed water by surfactant molecules. In order to evaluate the competitive adsorption of the water and organics on the same surface, a comparison of their adsorption energies is required.79 Although this comparison discards any reinforcement or debilitation of the organic molecule adsorption onto goethite due to interactions between them and surrounding water molecules, it provides a difference in energy for the hydrated surface and the molecule attached to the dry surface. This approach is further supported by evidence from molecular dynamics simulations showing no significant interaction between the methanoic acid adsorbed onto calcium fluoride and the surrounding liquid water, while the relative energies for the different adsorption sites were not affected by the addition of the solvent.68 To do this, the methodology established in previous studies was followed for the interaction of surfactants with different mineral surfaces, such as calcite,78,79 fluorite,77,79 scheelite58 and fluorapatite.46 It is clear from Table 2 that the adsorption energies of all three organic molecules on goethite surfaces are higher than the surface hydration energies. Thus from a thermodynamics perspective, these three organic molecules will be able to displace the water molecules on the goethite surfaces. Our results suggest that fulvic and humic acids could be eliminated from the aquifers via adsorption of their carboxylic ligands to goethite surfaces, such as those expected to be present in ZVI technologies.4. Conclusions
We have carried out a series of static energy minimisations to investigate the interactions between three organic molecules with diverse functional groups and a series of goethite surfaces. Our calculated results show that the strongest adsorption occurs when the adsorbate molecule is able to form multiple interactions with the surface ions, including hydrogen-bonds.Overall, the energetically most favourable interfacial system is the eclipsed hydroxamic acid on the goethite {100} surface. We noted that in the relaxed structures of the hydroxamic acid/goethite systems, the initial staggered conformation has rotated through the C–N bond to form the eclipsed conformation. It was also found that in the case of hydroxyethanal, the system with the largest adsorption energy occurs when hydroxyethanal in its staggered conformation interacts with the surface rather than the eclipsed conformer. It is interesting to note that in this case, the initial eclipsed conformation rotated through the C–C bond to form the staggered conformation at the surface.
Adsorption of all the organic molecules is exothermic, which indicates that these molecules are good surfactants. On thermodynamic grounds, competition with pre-adsorbed water should allow the organic molecules to replace water at the major surfaces of goethite and form high coverages of the surfactant molecules at the surfaces. Consequently, we consider that organic molecules that contain at least one carboxylic acid group or any of its modifications can be effectively removed from polluted aquifers by goethite.
We also studied how splitting the carboxylic acid into carbonyl and hydroxy groups attached to two separate carbon atoms on the surfactant molecules affected adsorption to goethite. This gave greater flexibility to the molecule, which therefore enhanced the interaction with the goethite surfaces, as the molecule can form interactions with the iron ions via the carbonyl group and hydrogen-bonds with the surface oxygen and hydroxide species. Accordingly, surfactants with the hydroxymethanamide or hydroxyethanal functional groups were able to adsorb more strongly to goethite surfaces and hence should be easier to separate from polluted aquifers than normal carboxylic acids.
Future work will focus on studying the effect of temperature through molecular dynamics simulations, as well as evaluating the adsorption energy of the organic surfactants on the mineral components of sand, to compare the capability of each iron wall component to collect the pollutants from contaminated aquifers.
Acknowledgements
We acknowledge the Engineering & Physical Sciences Research Council (grant No. EP/K009567/2) for funding. The authors also acknowledge the use of the UCL Legion High Performance Computing Facility (Legion@UCL), and associated support services, in the completion of this work. D. S.-C. thanks UCL for a Graduate Global Excellence Award and an Overseas Research Scholarship from the UCL Industrial Doctorate Centre in Molecular Modelling and Materials Science. N. H. d. L. is grateful to the Royal Society for the provision of an Industry Fellowship. All data created during this research is openly available from the University of Cardiff Research Portal at http://dx.doi.org/10.17035/d.2016.0008219810.References
- R. M. Cornell and U. Schwertmann, The Iron Oxides, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, FRG, 2nd edn, 2003 Search PubMed.
- S. L. S. Stipp, M. Hansen, R. Kristensen, M. F. Hochella, L. Bennedsen, K. Dideriksen, T. Balic-Zunic, D. Léonard and H.-J. Mathieu, Chem. Geol., 2002, 190, 321–337 CrossRef CAS.
- C. L. Chun, R. M. Hozalski and W. A. Arnold, Environ. Sci. Technol., 2005, 39, 8525–8532 CAS.
- P. J. Shea, T. A. Machacek and S. D. Comfort, Environ. Pollut., 2004, 132, 183–188 CAS.
- S. F. O'Hannesin and R. W. Gillham, Ground Water, 1998, 36, 164–170 Search PubMed.
- G. A. Waychunas, C. S. Kim and J. F. Banfield, J. Nanopart. Res., 2005, 7, 409–433 CrossRef CAS.
- S. R. Kanel, B. Manning, L. Charlet and H. Choi, Environ. Sci. Technol., 2005, 39, 1291–1298 CAS.
- X. Li and W. Zhang, Langmuir, 2006, 22, 4638–4642 CAS.
- J. A. Mielczarski, G. M. Atenas and E. Mielczarski, Appl. Catal., B, 2005, 56, 289–303 CrossRef CAS.
- Z. Chen, X. Jin, Z. Chen, M. Megharaj and R. Naidu, J. Colloid Interface Sci., 2011, 363, 601–607 CrossRef CAS PubMed.
- S. J. T. Pollard, M. Lythgo and R. Duarte-Davidson, in Assessment and Reclamation of Contaminated Land, ed. R. E. Hester and R. M. Harrison, The Royal Society of Chemistry, Cambridge, 2001, pp. 1–20 Search PubMed.
- W. Rulkens, R. Tichy and J. T. C. Grotenhuis, Water Sci. Technol., 1998, 37, 27–35 CAS.
- C. Nathanail and R. Bardos, Reclamation of Contaminated Land, Wiley, Chicester, 2004 Search PubMed.
- J. Scullion, Naturwissenschaften, 2006, 93, 51–65 CrossRef CAS PubMed.
- A. C. Singer, C. J. van der Gast and I. P. Thompson, Trends Biotechnol., 2005, 23, 74–77 CAS.
- T. Gentry, C. Rensing and I. Pepper, Crit. Rev. Environ. Sci. Technol., 2004, 34, 447–494 CrossRef CAS.
- T. Jong and D. L. Parry, Chemosphere, 2005, 60, 254–265 CAS.
- C. N. Mulligan, R. N. Yong and B. F. Gibbs, Eng. Geol., 2001, 60, 371–380 CrossRef.
- J. Aguilar, P. Bouza, C. Dorronsoro, E. Fernández, J. Fernández, I. García, F. Martín and M. Simón, Soil Use Manage., 2004, 20, 451–453 CrossRef.
- M. Shin, S. F. Barrington, W. D. Marshall and J.-W. Kim, Chemosphere, 2005, 58, 735–742 CAS.
- G. Park, H.-S. Shin and S.-O. Ko, J. Environ. Sci. Health, Part A: Toxic/Hazard. Subst. Environ. Eng., 2005, 40, 881–897 CAS.
- J. A. Adams and K. R. Reddy, Groundwater Monit. Rem., 2003, 23, 85–94 CAS.
- R. E. Saichek and K. R. Reddy, Crit. Rev. Environ. Sci. Technol., 2005, 35, 115–192 CrossRef CAS.
- R. Armishaw, R .P. Bardos, R. M. Dunn, J. M. Hill, M. Pearl, T. Rampling and P. A. Wood, Review of Innovative Contaminated Soil Clean-Up Processes, Warren Springs, Stevenage, 1992 Search PubMed.
- N. H. de Leeuw and T. G. Cooper, Geochim. Cosmochim. Acta, 2007, 71, 1655–1673 CrossRef CAS.
- K. D. Kwon and J. D. Kubicki, Langmuir, 2004, 20, 9249–9254 CAS.
- A. T. Stone, A. Torrents, J. Smolen, D. Vasudevan and J. Hadley, Environ. Sci. Technol., 1993, 27, 895–909 CAS.
- E. M. Cooper and D. Vasudevan, J. Colloid Interface Sci., 2009, 333, 85–96 CrossRef CAS PubMed.
- M. V. Biber and W. Stumm, Environ. Sci. Technol., 1994, 28, 763–768 CrossRef CAS PubMed.
- K. Hanna, S. Martin, F. Quilès and J.-F. Boily, Langmuir, 2014, 30, 6800–6807 CrossRef CAS PubMed.
- Y. S. Hwang, J. Liu, J. J. Lenhart and C. M. Hadad, J. Colloid Interface Sci., 2007, 307, 124–134 CrossRef CAS PubMed.
- M. Lindegren, J. S. Loring and P. Persson, Langmuir, 2009, 25, 10639–10647 CrossRef CAS PubMed.
- C. R. Evanko and D. A. Dzombak, J. Colloid Interface Sci., 1999, 214, 189–206 CrossRef CAS PubMed.
- L. S. Balistrieri and J. W. Murray, Geochim. Cosmochim. Acta, 1987, 51, 1151–1160 CrossRef CAS.
- M. A. Ali and D. A. Dzombak, Environ. Sci. Technol., 1996, 30, 1061–1071 CrossRef CAS.
- J.-F. Boily, P. Persson and S. Sjöberg, Geochim. Cosmochim. Acta, 2000, 64, 3453–3470 CrossRef CAS.
- C. F. Whitehead, R. F. Carbonaro and A. T. Stone, Aquat. Geochem., 2015, 21, 99–121 CrossRef CAS.
- T. A. Kendall, M. F. Hochella Jr and U. Becker, Chem. Geol., 2005, 216, 17–35 CrossRef CAS.
- C. R. Evanko and D. A. Dzombak, Environ. Sci. Technol., 1998, 32, 2846–2855 CrossRef CAS.
- K. Lalonde, A. Mucci, A. Ouellet and Y. Gélinas, Nature, 2012, 483, 198–200 CrossRef CAS PubMed.
- L. Weng, W. H. van Riemsdijk, L. K. Koopal and T. Hiemstra, Environ. Sci. Technol., 2006, 40, 7494–7500 CrossRef CAS PubMed.
- L. Weng, W. H. Van Riemsdijk and T. Hiemstra, J. Colloid Interface Sci., 2007, 314, 107–118 CrossRef CAS PubMed.
- S. Kang and B. Xing, Langmuir, 2008, 24, 2525–2531 CAS.
- J. Greathouse, K. Johnson and H. Greenwell, Minerals, 2014, 4, 519–540 CrossRef CAS.
- B. Gu, J. Schmitt, Z. Chen, L. Liang and J. F. McCarthy, Geochim. Cosmochim. Acta, 1995, 59, 219–229 CrossRef CAS.
- D. Mkhonto, P. E. Ngoepe, T. G. Cooper and N. H. de Leeuw, Phys. Chem. Miner., 2006, 33, 314–331 CrossRef CAS.
- G. W. Watson, E. T. Kelsey, N. H. de Leeuw, D. J. Harris and S. C. Parker, J. Chem. Soc., Faraday Trans., 1996, 92, 433–438 RSC.
- M. Born and K. Huang, Dynamical theory of crystal lattices, Oxford Classic Texts, Clarendon Press, Oxford, 1988 Search PubMed.
- D. Parry, Surf. Sci., 1975, 49, 433–440 CrossRef CAS.
- D. Parry, Surf. Sci., 1976, 54, 195 CrossRef.
- R. Fletcher and C. M. Reeves, Comput. J., 1964, 7, 149–154 CrossRef.
- M. J. Norgett and R. Fletcher, J. Phys. C: Solid State Phys., 1970, 3, L190–L192 CrossRef CAS.
- B. Dick and A. Overhauser, Phys. Rev., 1958, 112, 90–103 CrossRef.
- G. V. Lewis and C. R. A. Catlow, J. Phys. C: Solid State Phys., 1985, 18, 1149–1161 CrossRef CAS.
- P. S. Baram and S. C. Parker, Philosophical Magazine Part B, 1996, 73, 49–58 CrossRef CAS.
- K. Schröder, J. Sauer, M. Leslie, C. R. A. Catlow and J. M. Thomas, Chem. Phys. Lett., 1992, 188, 320–325 CrossRef.
- P. Dauber-Osguthorpe, V. A. Roberts, D. J. Osguthorpe, J. Wolff, M. Genest and A. T. Hagler, Proteins, 1988, 4, 31–47 CrossRef CAS PubMed.
- T. G. Cooper and N. H. de Leeuw, Langmuir, 2004, 20, 3984–3994 CrossRef CAS PubMed.
- T. G. Cooper and N. H. de Leeuw, Chem. Commun., 2002, 1502–1503 RSC.
- N. H. de Leeuw and S. C. Parker, Res. Chem. Intermed., 1999, 25, 195–211 CrossRef CAS.
- S. C. Parker, N. H. de Leeuw and S. E. Redfern, Faraday Discuss., 1999, 114, 381–393 RSC.
- N. H. de Leeuw, F. M. Higgins and S. C. Parker, J. Phys. Chem. B, 1999, 103, 1270–1277 CrossRef CAS.
- N. H. de Leeuw, Z. Du, J. Li, S. Yip and T. Zhu, Nano Lett., 2003, 3, 1347–1352 CrossRef CAS.
- Z. Du and N. H. de Leeuw, Surf. Sci., 2004, 554, 193–210 CrossRef CAS.
- B. Fubini, V. Bolis, M. Bailes and F. S. Stone, Solid State Ionics, 1989, 32–33, 258–272 CrossRef.
- N. H. de Leeuw and S. C. Parker, J. Phys. Chem. B, 1998, 102, 2914–2922 CrossRef CAS.
- P. Fenter, P. Geissbühler, E. DiMasi, G. Srajer, L. B. Sorensen and N. C. Sturchio, Geochim. Cosmochim. Acta, 2000, 64, 1221–1228 CrossRef CAS.
- N. H. de Leeuw and T. G. Cooper, J. Mater. Chem., 2003, 13, 93–101 RSC.
- N. Almora-Barrios and N. H. de Leeuw, Langmuir, 2010, 26, 14535–14542 CrossRef CAS PubMed.
- N. Almora-Barrios and N. H. De Leeuw, Cryst. Growth Des., 2012, 12, 756–763 CAS.
- N. Almora-Barrios and N. H. de Leeuw, CrystEngComm, 2010, 12, 960–967 RSC.
- N. Almora-Barrios, K. F. Austen and N. H. de Leeuw, Langmuir, 2009, 25, 5018–5025 CrossRef CAS PubMed.
- T. G. Cooper and N. H. de Leeuw, Surf. Sci., 2003, 531, 159–176 CrossRef CAS.
- J. L. Hazemann, A. Manceau, P. Sainctavit and C. Malgrange, Phys. Chem. Miner., 1992, 19, 25–38 CrossRef CAS.
- W. A. Deer, P. R. A. Howie and P. J. Zussman, An Introduction to the Rock-Forming Minerals, Prentice Hall, 1992 Search PubMed.
- J. Hur and M. A. Schlautman, J. Environ. Qual., 2004, 33, 1733–1742 CrossRef PubMed.
- T. G. Cooper and N. H. de Leeuw, J. Mater. Chem., 2004, 14, 1927 RSC.
- N. H. de Leeuw and T. G. Cooper, Cryst. Growth Des., 2004, 4, 123–133 CAS.
- N. H. de Leeuw, S. C. Parker and K. H. Rao, Langmuir, 1998, 14, 5900–5906 CrossRef CAS.
- X. Chen, H. Hu, J. Liu, H. Chen and Q. Wang, J. Cent. South Univ., 2015, 22, 1626–1634 CrossRef CAS.
- B. A. Holmén, M. I. Tejedor-Tejedor and W. H. Casey, Langmuir, 1997, 13, 2197–2206 CrossRef.
- B. A. Holmén and W. H. Casey, Geochim. Cosmochim. Acta, 1996, 60, 4403–4416 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Interatomic potentials for the interaction of methanoic acid, hydroxamic acid and hydroxyethanal with goethite were given in Tables ESI-1, ESI-2 and ESI-3. See DOI: 10.1039/c6ra12377e |
| This journal is © The Royal Society of Chemistry 2016 |