
Can single graphene nanodisks be used as Raman enhancement platforms?†
Abstract
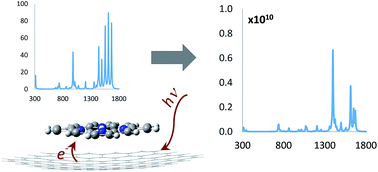
Recent works on plasmonic properties of graphene molecules have pointed out the possibility of building optical devices and Raman sensors using individual molecules. Here, the Raman spectra of different biomolecular units adsorbed on a zig-zag graphene nanodisk of ninety six carbon atoms (C96) are investigated using time-dependent perturbation methods. Static and pre-resonance conditions have been simulated to elucidate the Raman enhancement mechanism via ground state and excited state interactions with the nanodisk. Stacking and H-π complexes formed by the pyronine cation, porphine, tetrabenzoporphine and phthalocyanine with C96 have been considered. Static polarizability changes, charge transfer transitions, surface resonance, molecule-surface vibrational couplings and symmetry factors may all influence the Raman spectra of the molecules. We have explored the role played by each factor in the different conformational dispositions. Our results point out the use of small nanographene structures as promising for the development of SERS platforms at the frontier of nanometer and subnanometer scales.
 Please wait while we load your content...
Please wait while we load your content...

