
Structural transitions and electronic properties of sodium superoxide at high pressures†
Abstract
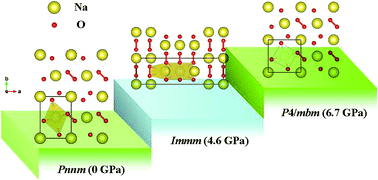
Sodium superoxide (NaO2) has attracted considerable attention as the main discharge product in Na–air batteries due to its specific energy, which exceeds that of the Li-ion battery. Pressure has become an irreplaceable tool to improve or alter the physical properties of a given material. By utilizing first-principles swarm structure-searching predictions, herein, we for the first time investigate the structures and electronic properties of NaO2 in the pressure range of 0–20 GPa. It is found that the orthorhombic Pnnm structure at ambient pressure transforms to another orthorhombic Immm structure at approximately 4.6 GPa, and subsequently to the tetragonal P4/mbm structure at 6.7 GPa. The pressure-induced structural transitions are mainly derived from the denser polyhedral packing and higher coordination number. It is interesting to find that the superoxide group (O2−) is maintained over the entire pressure range considered. Analysis of the electronic band structure and density of states shows that the structures found exhibit intriguing half-metallic magnetism. This study enables an opportunity to understand the structures and electronic properties of NaO2 at high pressures.
 Please wait while we load your content...
Please wait while we load your content...

