
Synthesis, structure, photophysical, electrochemical properties and antibacterial activity of brominated BODIPYs†
Abstract
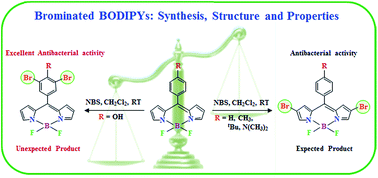
A series of mono- and di-brominated BODIPYs (1–5) was synthesized and characterized with a view to study the performance of dyes towards antibacterial activity. Regioselective bromination at the 2- and 2,6-positions of the BODIPY core was achieved with quantitative yield. The bromination of meso-(4-hydroxyphenyl) BODIPY (5) yielded an unexpected dibromo derivative, where the bromine groups were installed at the 3,5-positions of the phenyl ring rather than the 2,6-positions of the BODIPY core, which is confirmed and supported by UV-visible, fluorescence, and 1H NMR spectroscopic analyses, electrochemical studies, and also by single crystal X-ray crystallography. We observed a red shift of ∼16 nm in the absorption and 20–29 nm in the emission spectra in CH2Cl2 for the installation of each bromine group at the BODIPY core. The small difference between the first reduction potentials of the parent and dibromo derivative (5 and 5b) reveal that dibromination does not occur on the pyrrolic moiety. The intermolecular interactions involving C⋯H, F⋯H, H⋯H, and Br⋯H are the key factors in stabilizing the molecular crystal packing. The antibacterial properties of these dyes were investigated and the brominated derivatives showed better antibacterial effects than their corresponding parent BODIPYs, particularly the unusual dibromo derivative, 5b.
 Please wait while we load your content...
Please wait while we load your content...

