
Improved photovoltaic performance of mesoporous perovskite solar cells with hydrogenated TiO2: prolonged photoelectron lifetime and high separation efficiency of photoinduced charge†
Ting Su,
Yulin Yang*,
Guohua Dong,
Tengling Ye,
Yanxia Jiang and
Ruiqing Fan*
MIIT Key Laboratory of Critical Materials Technology for New Energy Conversion and Storage, School of Chemistry and Chemical Engineering, Harbin Institute of Technology, Harbin 150001, P. R. China. E-mail: ylyang@hit.edu.cn; fanruiqing@hit.edu.cn; Fax: +86-451-86418270
First published on 28th June 2016
Abstract
Hydrogenated titanium dioxide (H-TiO2) nanocrystals and nanorods (H-TNRs) are successfully synthesized and employed as electron transfer materials in mesoscopic perovskite solar cells (PSCs). In comparison with PSCs based on untreated TiO2, PSC devices based on H-TiO2 exhibit a significantly greater photovoltaic performance with a solar-to-electric energy conversion efficiency of over 13%. A 15.79% increase in Jsc (17.29 mA cm−2 to 20.02 mA cm−2) was observed in PSCs based on TiO2 and H-TiO2 nanopowders, and also there is a slight amplification of the open-circuit voltage (Voc) from 0.92 V to 0.97 V. The H-TiO2 nanocrystals exhibit a broader absorption band in the visible wavelength range, the donor density is increased and the band potential is shifted positively, which yield the enhanced driving force for electron injection thus elevating the current density of the PSCs. Moreover, it is elucidated that the electron behavior of the H-TiO2 nanocrystals can prolong the photogenerated charge lifetime, slow down the recombination rate of the electron–hole pairs and elevate the photoinduced charge separation efficiency through surface photovoltage spectroscopy (SPS) and transient photovoltage measurement (TPV).
1. Introduction
Recently, the discovery of organic–inorganic perovskite solar cells (PSCs) has offered promising routes for the development of low-cost and clean energy in the future. Organic–inorganic hybrid perovskite materials like CH3NH3PbI3 with a high light absorption coefficient, great and balanced charge carrier mobility, long charge diffusion lengths, and tunable bandgaps have been applied prevalently. By making unprecedented development, researchers have managed to reach solar cell efficiencies that have passed those of traditional photovoltaic solar cells. The power conversion efficiency of PSCs has been dramatically increased from 3.81% in 2009![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 1 to a certified value of 22.1% in 2016.2
1 to a certified value of 22.1% in 2016.2
Mesoporous PSCs are one kind of PSC that has drawn great attention in recent years. Although mesoporous PSCs are relatively complex to fabricate in comparison with planar heterojunction PSCs, mesoporous PSCs attract more attention due to unique advantages such as the scaffold layer function, little hysteresis phenomenon and the effective charge transport properties of mesoscopic metal oxide materials. Many methods have been employed to modify mesoporous electron transport materials in order to achieve high-performance photovoltaic devices, such as modifying the surface, morphology and structure,3–9 tuning the chemical or physical properties, enhancing the light harvesting abilities,10,11 and adjusting the electronic energy band structure.4,12,13 Among various oxides, TiO2 as the most ordinary semiconductor has been extensively used as an electron transfer layer material in mesoporous perovskite solar cells, and has been studied in a variety of applications including dye-sensitized solar cells, photocatalysts, and organic photovoltaic. Numerous methods have been investigated to enhance its visible light absorption with a large bandgap, adjust the morphology or electronic structure and improve the donor density and electrical conductivity. Rutile TiO2 nanorod-based perovskite solar cells have been fabricated by Xu and co-workers, showing a photoelectric conversion efficiency (PCE) of 11.7%.14 Qin et al. reported PSCs based on Au-decorated TiO2 nanofibers reaching a high PCE of 14.92%.6 Yue and co-workers reported PSCs based on a PMMA-mediated mesoporous TiO2 layer exhibiting a best PCE of ≥14%.15 However, all the reported modifications are relatively complicated, the separation efficiency of the photoinduced charge is still low, and the lifetime of the photogenerated charge is short, which hinder the photovoltaic performance to be further optimized. Hydrogenation treatment has emerged as an effective surface hydrogenation way to modify the energy band structure as well as the light harvesting properties of materials and it is feasible to induce dramatic structural changes in TiO2.16–22 Thus far, hydrogenation treatments are prevailing in photocatalysis, field-emission transistors and lithium batteries, whereas they have not been investigated in perovskite solar cells. Considering the great potential of hydrogenation treatment, incorporating hydrogenated TiO2 into PSCs should be thoroughly investigated. However, to the best of our knowledge, employing surface hydrogenation treatment in perovskite solar cells has been rarely studied.
Herein, we employ two kinds of TiO2 materials including hydrogenated titanium dioxide (H-TiO2) nanocrystals and nanorods (H-TNRs) as the hydrogenated TiO2 mesoporous layer for the PSCs. The highest efficiency of the solar cells based on the hydrogenated samples is effectively enhanced to ≥13%. Our work is mainly about the electron transfer and recombination behavior and elucidates the reason for the enhanced PCE. UV-vis spectroscopy and Mott–Schottky analysis are applied to look into the light absorbing ability, bandgap alteration of TiO2 and donor density variation after the hydrogen thermal treatment. Especially, the changed electron transfer behavior and the lifetime after hydrogenation were investigated by surface photovoltage spectroscopy (SPS) and transient photovoltage measurement (TPV) methods, respectively. These analyses give strong evidences to explain the enhanced short-circuit current density (Jsc) and PCE. Besides, PSCs based on H-TiO2 samples show more rapid electron transport and a longer electron lifetime than the untreated TiO2 samples according to other related electrochemical properties. Such an innovative surface-modified design reported in this work provides a new pathway to improve the performance of TiO2-based PSCs.
2. Results and discussion
2.1 Morphology and crystal structure analysis
X-ray diffraction patterns of the TiO2 and H-TiO2 samples are shown in Fig. 1a. From the patterns, both anatase (JCPDS # 21-1272) and rutile (JCPDS # 21-1276) phases were observed in the highly crystalline TiO2 and H-TiO2 samples. The two samples possessed almost the same patterns, indicating that no other phase appeared after the hydrogen thermal treatment. The peak intensity of the rutile (110) peak at 2θ = 27.4° in the H-TiO2 samples is stronger than that of the TiO2 samples, suggesting that the hydrogen thermal treatment had a tendency to induce the generation of the rutile phase. According to the equation χ = (1 + 0.8IA/IR)−1,23 the content of rutile by XRD (%) can be easily estimated from the integrated XRD peak intensity. The rutile content calculations of the TiO2 and H-TiO2 samples in our work gave 56.52% and 57.12%, respectively. Also, the H-TNR samples show almost the same patterns with the untreated samples. The SEM pictures of the H-TiO2 film morphology and H-TNR samples are displayed in Fig. 1b and c, respectively. No distinctive change occurred in the film morphology before and after the hydrogen thermal treatment. The H-TiO2 samples were mainly nano-sized spherical particles with a small amount of V-shaped TiO2 aggregated by nanoparticles. The low resolution TEM images of H-TiO2 are shown in Fig. 1d and e. The size of the TiO2 spherical particles was around 25 nm in diameter. And the length size of the V-shaped crystal was around 200 nm. The high resolution TEM images in Fig. 1d are used to identify the anatase and rutile phases, with the inter-planar spacings of 0.325 and 0.352 nm corresponding to the rutile phase (110) and anatase phase (101), respectively. It was difficult to distinguish the disordered layer of the H-TiO2 nanopowders in Fig. 1e due to the light treatment of hydrogenation at low temperature and at normal pressure. Fig. 1c and f show that the H-TNR samples had a diameter from 10–40 nm and the nanorods were mixed with 20 nm spherical nanoparticles. To reveal the surface chemical bonding of the TiO2 and H-TiO2 samples, the XPS analysis method was employed. As shown in Fig. S1,† the samples have no different features in the Ti 2p core level XPS spectra. There is no peak centered at 457.0 eV, which is the characteristic feature for Ti3+ ions. The results show that the hydrogenation treatment does not introduce Ti 2p1/2 peak variation. | ||
| Fig. 1 (a) X-ray diffraction patterns of TiO2 and H-TiO2 nanopowder, film morphology of (b) H-TiO2 mesoporous film and (c) H-TiO2 nanofibers, (d) TEM image of hydrogenated TiO2 treated at 300 °C, (e) high resolution TEM image of TiO2 correlating to the (101) and (110) inter-planar crystal spacing and (f) TEM image of H-TNR. | ||
PSCs were fabricated according to the experimental section. A cross-sectional SEM image for the H-TiO2 mesoporous PSCs is shown in Fig. 2a. The cross-sectional SEM image displays the structure of mesoporous PSCs. The compact TiO2 layer was spin-coated on FTO with a thickness of less than 100 nm, and the mesoporous electron transport layer was deposited on the TiO2 compact layer, and the perovskite material was spin-coated on the mesoporous TiO2 layer and part of the MAPbI3 permeated in the mesoporous TiO2 layer, and the thickness of the mesoporous TiO2 layer and MAPbI3 was about 600 nm. The mesoporous H-TiO2 nanoparticles functioned as the scaffold layer and as effective charge transport layer material. The hole transport material of spiro-OMeTAD was formed on the top of the perovskite layer, and at last a gold layer was deposited on the hole transport material to form the back electrode. The thickness of the HTM and the gold electrode layer was 200 nm and 100 nm, respectively. Fig. 2b shows the UV-vis spectra of the FTO/TiO2/MAPbI3 and FTO/H-TiO2/MAPbI3 films. After hydrogen thermal treatment, the H-TiO2 film system showed strong absorption in the visible region from 350–750 nm due to the modification of surface hydrogenation compared with the TiO2 film system. The XRD pattern of the TiO2/perovskite layer is shown in Fig. S2.†
 | ||
| Fig. 2 (a) Cross-sectional scanning electron microscopy image for the H-TiO2 mesoporous PSCs, and (b) UV-vis spectra of the FTO/TiO2/MAPbI3 and FTO/H-TiO2/MAPbI3 films. | ||
2.2 Electrical and electrochemical properties
The current density–voltage (J–V) curves for mesoporous PSCs were measured under simulated air mass (AM) 1.5 irradiance of 100 mW cm−2 and are shown in Fig. 3a. A mask was employed to unify the illuminated active area and further to avoid edge effects during measurements.24 The photovoltaic parameters obtained in the J–V measurements are tabulated in Table 1. As shown in Fig. 3a, a 15.79% increase in Jsc (17.29 mA cm−2 to 20.02 mA cm−2) was observed in PSCs based on TiO2 and H-TiO2 nanopowders, and the amplification of the open-circuit voltage (Voc) (0.92 V to 0.97 V) was slight and the fill factor (FF) (0.69 to 0.67) had little variation after hydrogen thermal treatment, indicating the great influence on the Jsc due to the surface hydrogenation. And PSCs based on TNR and H-TNR showed lower efficiencies compared with PSCs based on TiO2 nanopowders, with 7.84% and 9.52%, respectively. In order to certify the efficiency accuracy, mass parallel experiments were carried out and a PCE histogram of 40 devices based on TiO2 and H-TiO2 electron transport layers is shown in Fig. 3b confirming the high repeatability of the devices. In addition to the high Jsc, H-TiO2-based PSCs showed relatively little hysteresis in the J–V measurement, with a negligible change in Jsc with either direction of the voltage sweep (Fig. S4†). The results were in accordance with the improved photovoltaic performance of H-TiO2 mesoporous PSCs. The device architecture of the mesoporous PSCs based on TiO2 and H-TiO2 is shown in Fig. 3c, and the preparation details of the H-TiO2-based PSCs are described in the experimental section. The mesoporous PSC structure can be obviously distinguished from the abbreviated drawing. Besides, the energy level diagram of the device including the TiO2, MAPbI3 and spiro-OMeTAD film system is shown in Fig. 3d. | ||
| Fig. 3 (a) J–V curves of the PSCs based on TiO2 and H-TiO2, (b) PCE histogram of 40 devices based on H-TiO2 electron transport layer, (c) device architecture of mesoporous PSC based on TiO2 and H-TiO2, and (d) energy level diagram of the device. | ||
| Sample | Jsc (mA cm−2) | Voc (V) | FF | η (%) |
|---|---|---|---|---|
| TiO2 | 17.29 ± 1.20 | 0.92 ± 0.02 | 0.69 ± 0.03 | 10.84 ± 0.80 |
| H-TiO2 | 20.02 ± 1.60 | 0.97 ± 0.04 | 0.68 ± 0.02 | 13.22 ± 1.00 |
| TNR | 13.96 ± 1.00 | 0.85 ± 0.04 | 0.66 ± 0.02 | 7.84 ± 0.60 |
| H-TNR | 15.44 ± 1.40 | 0.88 ± 0.03 | 0.69 ± 0.02 | 9.52 ± 0.80 |
The IPCE spectra of PSCs based on the TiO2 and H-TiO2 mesoscopic electron transport layer are shown in Fig. 4. The two kinds of PSCs displayed similar IPCE spectra profiles and high IPCE values in the wavelength range between 350 nm and 750 nm. Obviously, the PSCs based on the H-TiO2 mesoporous electron transport layer displayed a higher IPCE value of 79.3%, reflecting a more efficient photoinduced charge carrier separation and transport. The value of the PSCs based on H-TiO2 had a 12.83% enhancement compared with that of the PSCs based on untreated TiO2, which was consistent with Jsc. The slightly lower integrated current Jsc values of the IPCE measurements may be due to device degradation during the measurements or the spectral mismatch factor.
 | ||
| Fig. 4 IPCE spectra of PSCs based on TiO2 and H-TiO2 in the wavelength range from 300 nm to 850 nm. | ||
The enhanced photovoltaic performance can be mainly ascribed to the elevated short-circuit current density due to the variation of electronic structure and light absorption ability. In order to study the light absorption ability and band edge values of the TiO2 and H-TiO2 samples, UV-visible absorption and Mott–Schottky analyses were carried out. As shown in Fig. 5a, the hydrogen thermal treatments did affect the light absorption properties of TiO2. A strong UV light harvesting ability at around 400 nm originated from anatase TiO2. In comparison with untreated TiO2, the absorption band of the H-TiO2 samples was slightly red-shifted and showed light absorption at 400–700 nm, and almost utilized the whole visible light wavelength range. This result is in accordance with the colour variation from white to light yellow (inset of Fig. 5a). A relationship between the absorption coefficient (α) near the absorption edge and the optical bandgap (Eg) can be elucidated by the Tauc’s plot equation. From the Tauc’s plot (αhν)2 = A(hν − Eg), the bandgaps of the TiO2 and H-TiO2 nanopowders can be estimated where α is the absorption coefficient, h is Planck’s constant, ν means the light vibrational frequency, A represents a constant, and Eg is the bandgap.25 Tauc’s plots are shown in the inset of Fig. 5a. The bandgap value of H-TiO2 after hydrogenation for 30 min was 2.83 eV, which was narrower than that of untreated TiO2 (3.06 eV). The hydrogen thermal treatment can change Eg as well as increase the absorption of H-TiO2 samples in the visible light range. Hydrogenation can generate dangling bonds and a disordered surface layer of the TiO2 samples, which is feasible to introduce mid-gap electronic states. The narrowed bandgap originated from the existence of mid-gap electronic states in the samples according to our early study and former reports,17 and these mid-gap states may enhance the photovoltaic performance. As shown in Fig. 5a, the synthesized TiO2 nanocrystals showed a white colour, and after hydrogenation for 30 min, the white TiO2 nanocrystals turn light yellow. The TNR samples showed similar results and H-TNR had a darker colour compared with TNR. The light absorption was weaker compared with the TiO2 nanocrystals and the bandgap value of H-TNR was 2.91 eV.
 | ||
| Fig. 5 (a) UV-vis spectra of TiO2 and H-TiO2 nanopowders and (b) TNR and H-TNR samples. The inset shows the bandgap calculation by Tauc’s plot and a photo comparing the untreated white TiO2 and disordered engineered H-TiO2 nanocrystals. | ||
To further elucidate the electronic properties including the flat band potential (Ef) and the shift of the conduction band minimum in the TiO2 and the H-TiO2 samples, we carried out Mott–Schottky analysis. As shown in Fig. 6a, both semiconductor samples exhibited positive slopes and can be identified as n-type semiconductors, indicating that the hydrogen thermal treatment cannot change the type of semiconductor in our work. Besides the justification of the semiconductor type, we can also estimate donor densities according to the Mott–Schottky relationship:26
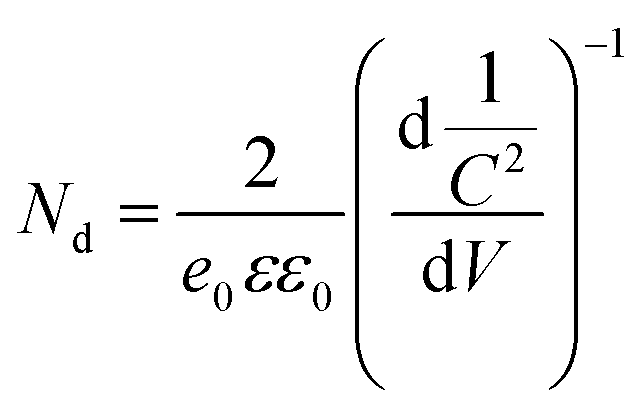
 | ||
| Fig. 6 Mott–Schottky plots collected in the dark for the TiO2 and the H-TiO2 films. | ||
In the Mott–Schottky relationship,  stands for the slope of the plots, since
stands for the slope of the plots, since  is constant, and Nd can be calculated from the equation. As shown in Fig. 6, the plot of the H-TiO2 sample displays a smaller slope compared with that of the TiO2 sample, suggesting a higher donor density or a higher electron concentration in H-TiO2. According to the equation above, the donor densities of the TiO2 and H-TiO2 nanopowders were calculated to be 2.79 × 1020 and 4.88 × 1020 cm−3, respectively. The enhanced donor density in H-TiO2 could improve the charge transport in the TiO2 nanoparticles, resulting in an enhanced charge collection efficiency. The Mott–Schottky relationship was not the absolute accurate approach to calculate the donor density, but the relative values can lead to a qualitative comparison that the H-TiO2 sample displayed a higher donor density after hydrogenation treatment. The increased donor density improved the charge transport in the semiconductor itself as well as the electron transport between the mesoporous electron transport layer and conducting TiO2 layer. The enhanced donor density could be the reason of the improvement in the photovoltaic performance.
is constant, and Nd can be calculated from the equation. As shown in Fig. 6, the plot of the H-TiO2 sample displays a smaller slope compared with that of the TiO2 sample, suggesting a higher donor density or a higher electron concentration in H-TiO2. According to the equation above, the donor densities of the TiO2 and H-TiO2 nanopowders were calculated to be 2.79 × 1020 and 4.88 × 1020 cm−3, respectively. The enhanced donor density in H-TiO2 could improve the charge transport in the TiO2 nanoparticles, resulting in an enhanced charge collection efficiency. The Mott–Schottky relationship was not the absolute accurate approach to calculate the donor density, but the relative values can lead to a qualitative comparison that the H-TiO2 sample displayed a higher donor density after hydrogenation treatment. The increased donor density improved the charge transport in the semiconductor itself as well as the electron transport between the mesoporous electron transport layer and conducting TiO2 layer. The enhanced donor density could be the reason of the improvement in the photovoltaic performance.
Mott–Schottky analyses also can be used to estimate the Ef location in order to confirm the shift of the conduction band minimum. After hydrogen thermal treatment, as shown in Fig. 6, linear extrapolation of the plots to the baselines can be applied to estimate Ef of the samples. The Ef value of the TiO2 film was −0.59 V (vs. SCE), whereas the Ef value of the H-TiO2 films was around −0.55 V (vs. SCE). The positive shift of the flat band increased the energy gap between the light absorption materials CH3NH3PbI3 and the conduction band of TiO2. The increased electron density was responsible for the positive shift of the Fermi energy level of TiO2 toward the conduction band, which facilitated the charge separation at the TiO2/electrolyte interface, by increasing the band bending at the surface of TiO2.27 The positive shift of the Fermi energy level of TiO2 can lead to a stronger driving force of photoinduced electron injection. As shown in Fig. 3d, under illumination, the perovskite materials like MAPbI3 absorbed light and generated electron–hole pairs. Thereafter, the electrons were injected into the mesoporous TiO2 layer and the holes were transported to the spiro-OMeTAD hole transport layer and Au back contact. Due to the narrowed bandgap of H-TiO2, it is more efficient for the electron injection to occur from the perovskite to the conduction band of H-TiO2 with an improved driving force, subsequently separating electrons from holes and reducing the probability of recombination.28 Compared with PSCs based on untreated TiO2, an elevated Jsc value can be obtained from PSCs based on H-TiO2. Based on the results mentioned, the enhancement can be ascribed to the stronger light absorption, the effective photoinduced charge transport and the prolonged lifetime of H-TiO2. Besides, the flat band of TiO2 after hydrogenation treatment was positively shifted suggesting that the electron injection process was more energetically favourable. Hydrogenation can also improve the electron transport/collection and restrain the recombination of photoinduced charge by providing a high conductive path and better interface contact. The PSCs based on TNR and H-TNR showing lower efficiencies can be ascribed to two reasons. On one hand, the light absorption in the UV-visible range was weaker and the bandgap value was a little bigger than that of the TiO2 nanopowders. On the other hand, the nanorod structure of TiO2 is not conducive for the electron transfer process between the perovskite layer and ETL due to the lower surface area and non-compact structure of the nanorods compared with the nanoparticles.
Furthermore, the electron recombination kinetics in different PSCs was characterized by the OCVD technique, which monitored the decay of Voc after turning off the illumination. Since it was measured in the dark, only the electrons recombined were revealed. Fig. 7a shows the OCVD curves of PSCs with TiO2 and H-TiO2. It was obvious that after 10 s in the dark under open-circuit conditions, the Voc of the PSCs decayed rapidly due to the internal trapping and de-trapping, and the Voc value of the PSC based on TiO2 decayed dramatically to 0 V after 15 s while the Voc value of the PSC based on H-TiO2 decayed slowly after 40 s. The PSCs based on H-TiO2 showed a higher onset voltage compared with the PSCs based on TiO2. The OCVD curve of the PSC based on H-TiO2 displayed a slower decay rate suggesting an inhibiting effect on the charge carrier recombination. The electron lifetime (τn) versus voltage curves are shown in Fig. 7b, and these electron lifetimes of the PSCs can be calculated using the following equation:  , where kB and T are the Boltzmann constant and the temperature, respectively. It was evident that the electron lifetime of the PSCs based on H-TiO2 was longer than that based on TiO2, which indicated that the charge recombination rate from the internal trapping and de-trapping of PSCs based on H-TiO2 was much lower versus PSCs based on TiO2.
, where kB and T are the Boltzmann constant and the temperature, respectively. It was evident that the electron lifetime of the PSCs based on H-TiO2 was longer than that based on TiO2, which indicated that the charge recombination rate from the internal trapping and de-trapping of PSCs based on H-TiO2 was much lower versus PSCs based on TiO2.
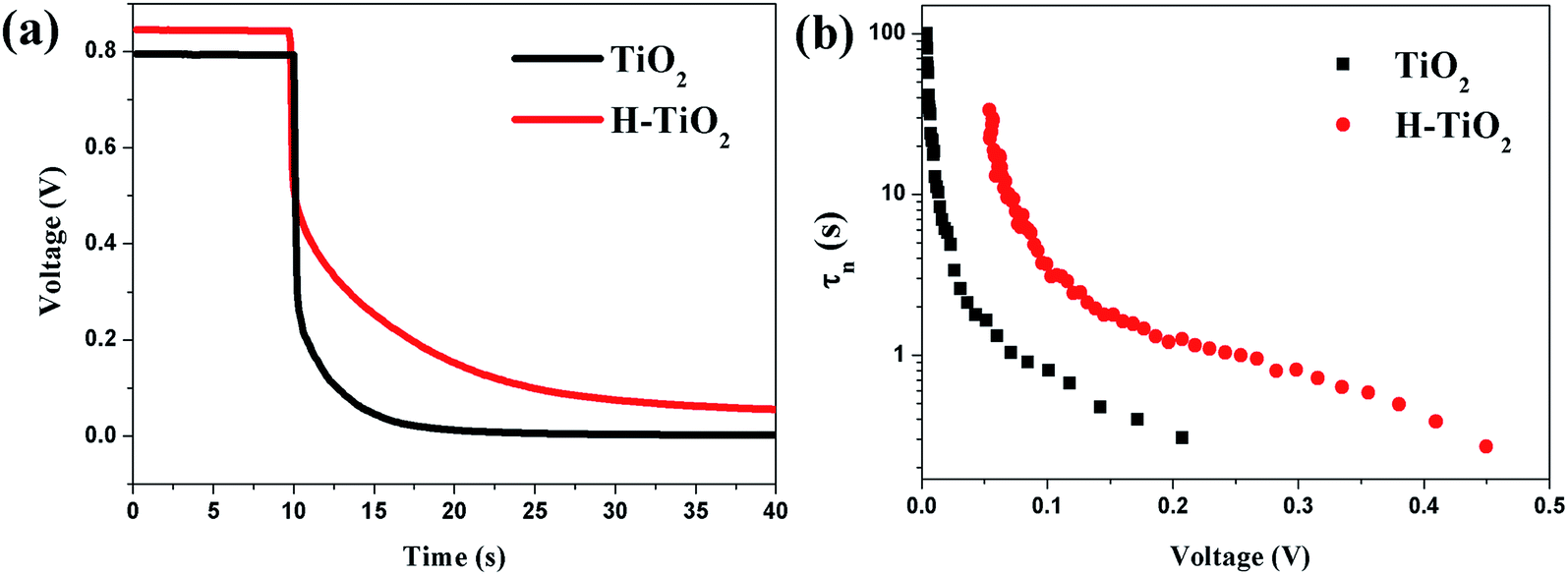 | ||
| Fig. 7 (a) The OCVD curves and (b) the electron lifetime as a function of Voc in the PSCs based on TiO2 and H-TiO2. | ||
Impedance spectroscopy (IS) measurements as a steady state method were utilized to analyze the carrier transport process in an electrochemical device, and current response measurements such as electron lifetime, transport resistance and chemical capacitance under the application of an AC voltage at different frequencies can be explained by IS measurement. The Nyquist plot shown in Fig. 8a mainly has two RC elements corresponding to the charge transfer process; the low frequency component was associated with the back contact between Au and the hole transport material (HTM) like the spiro-OMeTAD interface, and the high frequency component corresponded to charge transfer at the TiO2/perovskite/HTM interfaces.29–31 Under illumination and open-circuit voltage conditions, as shown in Fig. 8a, after hydrogenation thermal treatment, the mesoporous PSCs displayed a small semicircle in the low frequency region, which meant that a smaller charge transfer resistance existed between the TiO2 and perovskite interface and led to a lower series resistance of the device, indicating faster electron transport at the interface of the PSCs based on H-TiO2 as a mesoporous electron transport layer. This result was consistent with the shape of the J–V curves as well as with the differences of Voc observed in Fig. 3a.
 | ||
| Fig. 8 (a) The Nyquist plot of PSCs based on TiO2 and H-TiO2, and (b) PL quenching behaviors of films: glass/TiO2/CH3NH3PbI3 and glass/H-TiO2/CH3NH3PbI3 system with excitation at 425 nm. | ||
Since PL emission results from the recombination of free carriers, the measurement of PL emission was performed to reveal the behavior of photogenerated electrons and holes in semiconductors.32 Fig. 8b shows the PL spectra that were carried out with films: the glass/TiO2/CH3NH3PbI3 and H-TiO2/perovskite system with an excitation wavelength of 425 nm. The photoluminescence in the steady state was recorded to certificate the behavior of photogenerated electrons and holes of H-TiO2 nanopowders. As shown in Fig. 8b, the steady state PL intensity of CH3NH3PbI3 was greatly quenched to 54.1% of the original PL intensity of the glass/TiO2/CH3NH3PbI3 film compared with the glass/H-TiO2/CH3NH3PbI3 film. The strong fluorescence quenching was expected to originate from the charge carrier extraction across the interface.33,34 There was effective electron transport from CH3NH3PbI3 to TiO2 films and the photoinduced electron and hole in the perovskite layer could be separated efficiently after surface hydrogenation. The efficient quenching of the photoinduced charges of H-TiO2 on the perovskite layer could enhance the photoelectrical conversion efficiency.
Surface photovoltage spectroscopy (SPS) of TiO2 and the H-TiO2 nanopowder is shown in Fig. 9a. The SPS method is a well established non-destructive monitoring method to provide semiconductor information on things like surface band bending, the surface state and surface/bulk carrier recombination, and it is mainly sensitive to the electron transition-related process and subsequent separation. In particular, the application of the surface photovoltage response intensity is related to the photovoltaic activity by analyzing properties such as the charge separation efficiency of photoinduced carriers under illumination. Thus, according to the SPS principles, the stronger the SPS response intensity is, the higher the photoinduced charge separation efficiency is. As shown in Fig. 9a, the SPS response is in the range of 310 to 400 nm, which can be attributed to the transition from the valence band to the conduction band. In contrast with TiO2, the H-TiO2 samples displayed stronger SPV responses, presenting that H-TiO2 samples had a higher separation efficiency of photoinduced charges than that of TiO2 and this was beneficial for the PSCs as the electron transport layer. Furthermore, the SPS response was a little red-shifted and showed a relatively extended response wavelength range, resulting from the enhancement in absorbance of H-TiO2 after the hydrogenation treatment.
 | ||
| Fig. 9 (a) SPS of TiO2 and H-TiO2 samples and (b) TPV spectra of TiO2 and H-TiO2 samples, excited by a laser pulse of 355 nm and 100 μJ. | ||
The transient photovoltage measurement (TPV) technique was used to further investigate the differences in the dynamic properties like recombination and separation of photoinduced charge carriers in the TiO2 and H-TiO2 samples. As shown in Fig. 9b, there are two separation processes from the TPV curves: the fast carrier separation (<10−7 s) within one nanoparticle and inter-particle diffusion of photogenerated charge carriers at a long timescale.35 When the TiO2 and H-TiO2 samples were exposed to the laser pulse, positive TPV responses were observed for the two samples. From the moving direction of the photoinduced carriers it can be deduced that a positive response meant that the surface potential is positive compared with the bottom potential of the material. This result showed that the electron–hole pairs generated in the excited TiO2 could be separated efficiently, thus improving the photovoltaic response of H-TiO2.35,36 Meanwhile, from Fig. 9b, the electron–hole pair lifetime of the TPV responses in the H-TiO2 sample was prolonged from ∼10−5 s to ∼10−4 s. A longer lifetime leads to a slower recombination rate of the electron–hole pair. Consequently, it can be estimated that the obviously strengthened surface photovoltage response and the prolonged lifetime of the photogenerated charge could lead to a high photovoltaic performance. When the samples were employed as mesoporous transport layer materials in PSC devices, the perovskite layer injected electrons into the conduction band of the H-TiO2 mesoporous transport layer, which could accelerate the electron transport compared with the untreated TiO2 mesoporous transport layer therefore enhancing the photovoltaic performance. The results were consistent with the long lifetime and slower recombination rate of the electron–hole pair from OCVD analysis and dark J–V curves which also could explain the high efficiency of the PSCs.
The excellent photoelectrochemical properties of the PSCs of the as-prepared H-TiO2 samples could be attributed to three reasons. First, the unique morphology of the as-prepared H-TiO2 samples could afford a better scaffold layer function. The nano-sized spherical particles with a small amount of V-shaped crystals not only provide a faster process of electron transfer but also block the interface contact of the HTM layer and electron transfer layer. Second, the improved electron transition-related process and subsequent separation led to an energetically favourable electron injection process. It is known that the PEC performance of the solar cells is determined by charge separation and transfer process. In our experiment, the SPS and TPV analyses showed a stronger SPS response and a positive TPV response of the H-TiO2 samples, representing a high separation efficiency of photoinduced charges and efficient separation of electron–hole pairs generated in the excited TiO2, thus improving the photovoltaic response of H-TiO2. Meanwhile, a prolonged electron–hole pair lifetime from the TPV responses in the H-TiO2 sample could also lead to a slower recombination rate of the electron–hole pair. Third, the narrowed bandgap, the positive shift of the Fermi energy level of TiO2, and the enhanced donor density directly affected the valence band maximum and conduction band minimum leading to a stronger driving force of photoinduced electron injection. The result was closely related to the existence of oxygen vacancies after hydrogenation treatment and the existence of oxygen vacancies was certificated by EPR analysis as shown in Fig. S5.† The oxygen vacancies acted as shallow donors altering Ef of the H-TiO2 samples and were responsible for the enhanced donor density (Fig. 6). Also the introduced oxygen vacancy will improve the conductivity and carrier mobility, which could boost the injected electron transportation in H-TiO2. Consequently, the three reasons concluded from the characteristics of the H-TiO2 samples could elucidate the high photovoltaic performance of the PSCs.
3. Conclusions
In conclusion, H-TiO2 nanopowders and H-TNR samples have been successfully synthesized through a facile treatment of mixed gas flow and successfully applied into PSCs. PSCs based on H-TiO2 nanocrystals exhibit a notable enhancement of Jsc from 17.29 mA cm−2 to 20.02 mA cm−2 and of η from 10.84% to 13.22%. Surface hydrogenation leads to narrowed bandgap characteristics in the particles providing the possibility of high-performance PSC materials. The donor density is boosted and the occurrence of a flat band below the conduction band elucidates the possibility of applications in solar cells. The hydrogenation treatment cannot only facilitate the charge transport process, but also prolong the lifetime of photoinduced electrons and reduce electron recombination. The photovoltaic performance is obviously improved after the introduction of hydrogen thermal treatment, indicating that the hydrogenated TiO2 can be a good candidate for mesoporous electron transport materials of PSCs, inspiring the design of appropriate electron transport materials of PSCs.4. Experimental section
4.1 Preparation of hydrogenated TiO2 paste
The TiO2 nanopowders were prepared via a facile sol–gel hydrothermal method similar to our previous work.37 2 g organic template Pluronic F127 (Sigma-Aldrich, used as received) was added in 60 mL deionized water in a conical flask with vigorous stirring, and 0.4 mL HNO3 was then added in to the solution. 0.03 M titanium isopropoxide (98%, Alfa Aesar) was added drop-wise into the solution over the course of several minutes while vigorously stirring solution. This step gave a white precipitate. After accomplishing this step, the mixture was heated to 90 °C for about 5 h to obtain a gel-like mixture. The obtained mixture was almost translucent and was then transferred into a sealed Teflon-lined autoclave at 200 °C for 12 h. After cooling to room temperature, the resultant product was washed with DI water and absolute ethanol three times, and then dried at 80 °C for 6 h and subsequently calcined at 500 °C for 30 min to obtain the white TiO2 nanopowders. The synthesis of TiO2 nanorods were made using a modified hydrothermal process. 2 g P25 nanopowder (commercial Degussa P25 nanoparticles) was mixed with 10 mL 15 M NaOH aqueous solution with vigorous stirring for 30 min. The suspension was then transferred into a 20 mL Teflon-lined autoclave at 170 °C for 48 h. The white precipitates were washed with dilute HCl and water three times. The washed precipitates were calcined at 500 °C for 15 min to get the final product. The obtained TiO2 samples were placed in the quartz boat and heated in a tube furnace under a 400 sccm mixture gas flow of 10% H2 and 90% N2 for 30 min at 300 °C. The heating of the hydrogen thermal treatments was stepped at 1 °C min−1. The sample powders were maintained for stabilization in a vacuum for 1 h after hydrogen thermal treatment. TiO2 paste worked as an electron transport layer and was prepared by mixing TiO2 nanopowders, ethylcellulose (99.8%, Acros Organics), and α-terpinol in a certain amount of absolute ethanol (wt% ratio 1.68:0.45:7.87:30) with stirring and ultrasonic dispersion for 48 h.
4.2 PSC fabrication
The fabrication of the PSCs is in accordance with the literature38 and the details are as follows. Fluorine-doped tin oxide conducting glass (FTO, 90% transmittance in the visible region, 15 Ω per square, purchased from NSG, Japan) was used as substrates for PSCs. Before use, FTO glass was etched using Zn and 3 M hydrochloric acid aqueous solution. FTO glass was first washed with mild detergent, rinsed with distilled water several times and subsequently with acetone and isopropyl alcohol in an ultrasonic bath, finally dried under N2 stream, and treated in a UV ozone (UVO) cleaner for 10 min. To prepare the compact TiO2 layer (C-TiO2), 350 μL titanium isopropoxide (99.9% Sigma-Aldrich) was added drop-wise into 2.5 mL isopropyl alcohol (99.8%, Acros Organics). The compact TiO2 layers were spin-coated with a spin rate at 3000 rpm for 45 s on the FTO and calcined at 500 °C for 30 min. After the treatment, the films were immersed into 40 mM TiCl4 at 70 °C for 30 min and sintered at 500 °C for 30 min. Mesoporous TiO2 layers prepared by mixing TiO2/H-TiO2 with ethylcellulose, α-terpinol and ethanol were subsequently deposited using spin-coating with a spin rate of 5000 rpm for 45 s, and were also sintered at 500 °C for 30 min. For the preparation of PbI2 solution, 462 mg PbI2 was dissolved in 1 mL anhydrous DMF by stirring and then heated at 75 °C for 12 h. The CH3NH2I solution was obtained by dissolving 9 mg MAI into 1 mL 2-propanol and stirred for 2 h. The CH3NH3PbI3 layer was deposited by a modified two-step deposition method as described previously.39 PbI2 solution was spin-coated onto the TiO2 film at 3000 rpm for 5 s, and at 6000 rpm for 5 s (without loading time). After the deposition, the film was dried at 50 °C for 5 min and then at 100 °C for 5 min. After cooling to room temperature, the film was spin-coated with the CH3NH2I solution at 4000 rpm for 20 s. After being dried at 50 °C for 5 min and then at 100 °C for 5 min, the films were cooled down. Then, the hole transport layer solution was formed from the following steps. 1 mL chlorobenzene (99.8%, Acros Organics) solution containing 80 mg spiro-OMeTAD (99.8%, Toronto Research Chemicals), 28.5 μL tert-butylpyridine (TBP, 96%, Sigma-Aldrich), and 17.5 μL lithium bis(trifluoromethylsulfonyl)imide salt (Li-TFSI, 99%, Acros Organics, 520 mg in 1 mL acetonitrile) was used as the hole transport layer solution to form an electrolyte layer. The hole transport layer solution was spin-coated on a prepared film at 4000 rpm for 20 s. Finally, the samples were placed inside a desiccator overnight for adequate oxidation, and a gold layer (80 nm) was thermally deposited using an evaporator on the top of the hole transport layer to seal the device. In the experiments all of the chemical agents and solvents used were reagent grade without further purification.4.3 Solar cell characterization
The X-ray powder diffraction (XRD) measurement was analyzed with Cu Kα radiation using a Shimadzu XRD-6000 X-ray Diffraction instrument. Scanning electron microscopy (SEM) was performed using Rili SU 8000HSD Series Hitachi New Generation Cold Field Emission SEM. X-ray photoelectron spectroscopy (XPS) was recorded with an ESCALAB-250 spectrometer (Thermo, America) applying an Al Kα source under an ultra-high vacuum (3.5 × 10−7 Pa). UV-visible absorption spectra of TiO2 and H-TiO2 nanopowders were recorded on a Japan Shimadzu model UV-2250 spectrophotometer. The Mott–Schottky analysis was performed at f = 200 Hz with a small amplitude of 10 mV by a CHI660D electrochemical potentiostat using a three-electrode cell with a platinum wire as the counter electrode and a standard Ag/AgCl reference electrode. The supporting electrolyte used in the Mott–Schottky analysis was 0.01 M LiClO4, 0.1 M LiI and 1 mM I2. The surface photovoltage spectroscopy (SPS) analysis was carried on an instrument assembled by Jilin University. A xenon lamp (500 W) through a double-prism monochromator (SBP300, China) was used to obtain monochromatic light by passing light, and the lock-in amplifier (SR830, Stanford) can collect the signals. Transient photovoltage measurement (TPV) was investigated on a device and the samples were excited with a third harmonic Nd:YAG laser (Polaris II, New Wave Research, Inc.) with a 355 nm wavelength (50 μJ) and a pulse width of 5 ns. The obtained TPV signals were recorded with a digital phosphor oscilloscope (500 MHz, TDS 5054, Tektronix). An AAA Class 150 W solar simulator (SAN-EI ELECTRIC, model XES-40S2-CE, Japan) was employed to investigate the photovoltaic properties. The AM 1.5 global sunlight intensity was calibrated by a Newport Oriel PV reference cell and meter (model 91150 V). Photocurrent–photovoltage (I–V) curves were recorded by applying an external bias to the cell by a CHI660D electrochemical potentiostat. The scan rate was 10 mV s−1 and the delay time was 40 ms. Impedance spectroscopy (IS) measurements were recorded using a CHI660D electrochemical potentiostat. The measurements were obtained in the frequency range from 0.1 to 100 kHz in the dark. The incident photon-to-current efficiency (IPCE) spectra were conducted by IPCE kit equipment (model 2931-C, Newport, USA) a 1/4 m monochromator (model 74125 Oriel Cornerstone 260, Newport, USA), a 300 W xenon lamp (model 66902, Newport, USA), and a calibrated silicon detector (model 71675, Newport, USA). EPR spectra were recorded at 300 K with a Bruker ER083CS spectrometer using a microwave frequency of 9.85 GHz. The solid state photoluminescence was recorded on an FLS920 fluorescence spectrometer (Edinburgh) in the range of 500–850 nm at room temperature under an excitation wavelength of 425 nm that passed through a 650 nm low-pass filter. An Edinburgh Xe900 450 W xenon arc lamp was used as the excitation light source.Acknowledgements
This work was supported by National Natural Science Foundation of China (Grant 21571042, 21171044, 21371040 and 51502058), the National key Basic Research Program of China (973 Program, No. 2013CB632900).References
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef CAS PubMed.
- Research Cell Efficiency Records, http://www.nrel.gov/ncpv/images/efficiency_chart.jpg, accessed 28/05, 2015.
- H. S. Kim, J. W. Lee, N. Yantara, P. P. Boix, S. A. Kulkarni, S. Mhaisalkar, M. Grätzel and N. G. Park, Nano Lett., 2013, 13, 2412–2417 CrossRef CAS PubMed.
- P. Qin, A. L. Domanski, A. K. Chandiran, R. Berger, H. J. Butt, M. I. Dar, T. Moehl, N. Tetreault, P. Gao, S. Ahmad, M. K. Nazeeruddin and M. Grätzel, Nanoscale, 2014, 6, 1508–1514 RSC.
- M. H. Kumar, N. Yantara, S. Dharani, M. Grätzel, S. Mhaisalkar, P. P. Boix and N. Mathews, Chem. Commun., 2013, 49, 11089–11091 RSC.
- P. Qin, M. Paulose, M. I. Dar, T. Moehl, N. Arora, P. Gao, O. K. Varghese, M. Grätzel and M. K. Nazeeruddin, Small, 2015, 11, 5533–5539 CrossRef CAS PubMed.
- Y. Yue, T. Umeyama, Y. Kohara, H. Kashio, M. Itoh, S. Ito, E. Sivaniah and H. Imahori, J. Phys. Chem. C, 2015, 119, 22847–22854 CAS.
- S. S. Mali, C. S. Shim, H. Kim, P. S. Patil and C. K. Hong, Nanoscale, 2016, 8, 2664–2677 RSC.
- S. D. Sung, D. P. Ojha, J. S. You, J. Lee, J. Kim and W. I. Lee, Nanoscale, 2015, 7, 8898–8906 RSC.
- G. S. Han, H. S. Chung, B. J. Kim, D. H. Kim, J. W. Lee, B. S. Swain, K. Mahmood, J. S. Yoo, N.-G. Park, J. H. Lee and H. S. Jung, J. Mater. Chem. A, 2015, 3, 9160–9164 CAS.
- T. Liu, L. Liu, M. Hu, Y. Yang, L. Zhang, A. Mei and H. Han, J. Power Sources, 2015, 293, 533–538 CrossRef CAS.
- S. K. Pathak, A. Abate, P. Ruckdeschel, B. Roose, K. C. Godel, Y. Vaynzof, A. Santhala, S. I. Watanabe, D. J. Hollman, N. Noel, A. Sepe, U. Wiesner, R. Friend, H. J. Snaith and U. Steiner, Adv. Funct. Mater., 2014, 24, 6046–6055 CrossRef CAS.
- K. Manseki, T. Ikeya, A. Tamura, T. Ban, T. Sugiura and T. Yoshida, RSC Adv., 2014, 4, 9652–9655 RSC.
- Q. Jiang, X. Sheng, Y. Li, X. Feng and T. Xu, Chem. Commun., 2014, 50, 14720–14723 RSC.
- Y. Yue, T. Umeyama, Y. Kohara, H. Kashio, M. Itoh, S. Ito, E. Sivaniah and H. Imahori, J. Phys. Chem. C, 2015, 119, 22847–22854 CAS.
- Z. Lu, C.-T. Yip, L. Wang, H. Huang and L. Zhou, ChemPlusChem, 2012, 77, 991–1000 CrossRef CAS.
- A. Naldoni, M. Allieta, S. Santangelo, M. Marelli, F. Fabbri, S. Cappelli, C. L. Bianchi, R. Psaro and V. Dal Santo, J. Am. Chem. Soc., 2012, 134, 7600–7603 CrossRef CAS PubMed.
- G. C. Li, Z. H. Zhang, H. R. Peng and K. Z. Chen, RSC Adv., 2013, 3, 11507–11510 RSC.
- H. Zhou, Q. Chen, G. Li, S. Luo, T.-b. Song, H.-S. Duan, Z. Hong, J. You, Y. Liu and Y. Yang, Science, 2014, 345, 542–546 CrossRef CAS PubMed.
- Z. Wang, C. Y. Yang, T. Q. Lin, H. Yin, P. Chen, D. Y. Wan, F. F. Xu, F. Q. Huang, J. H. Lin, X. M. Xie and M. H. Jiang, Adv. Funct. Mater., 2013, 23, 5444–5450 CrossRef CAS.
- S. Lee, I.-S. Cho, J. H. Lee, D. H. Kim, D. W. Kim, J. Y. Kim, H. Shin, J.-K. Lee, H. S. Jung, N.-G. Park, K. Kim, M. J. Ko and K. S. Hong, Chem. Mater., 2010, 22, 1958–1965 CrossRef CAS.
- A. Kumar, R. Jose, K. Fujihara, J. Wang and S. Ramakrishna, Chem. Mater., 2007, 19, 6536–6542 CrossRef CAS.
- Y. Z. Li, Y. Fan and Y. Chen, J. Mater. Chem., 2002, 12, 1387–1390 RSC.
- M. A. Green, A. Ho-Baillie and H. J. Snaith, Nat. Photonics, 2014, 7, 506–514 CrossRef.
- J. Tauc, R. Grigorovici and A. Vancu, Phys. Status Solidi B, 1966, 15, 627–637 CrossRef CAS.
- F. Fabregat-Santiago, G. Garcia-Belmonte, J. Bisquert, P. Bogdanoff and A. Zaban, J. Electrochem. Soc., 2003, 150, E293 CrossRef CAS.
- D. Cronemeyer, Phys. Rev., 1959, 113, 1222–1226 CrossRef CAS.
- D. C. Hurum, A. G. Agrios, K. A. Gray, T. Rajh and M. C. Thurnauer, J. Phys. Chem. B, 2003, 107, 4545–4549 CrossRef CAS.
- B. Suarez, V. Gonzalez-Pedro, T. S. Ripolles, R. S. Sanchez, L. Otero and I. Mora-Sero, J. Phys. Chem. Lett., 2014, 5, 1628–1635 CrossRef CAS PubMed.
- A. Dualeh, T. Moehl, N. Tétreault, J. Teuscher, P. Gao, M. Nazeeruddin and M. Grätzel, ACS Nano, 2014, 8, 362–373 CrossRef CAS PubMed.
- J. A. Christians, R. C. Fung and P. V. Kamat, J. Am. Chem. Soc., 2014, 136, 758–764 CrossRef CAS PubMed.
- A. Ramchiary and S. K. Samdarshi, Chem. Phys. Lett., 2014, 597, 63–68 CrossRef CAS.
- S. D. Stranks, G. E. Eperon, G. Grancini, C. Menelaou, M. J. P. Alcocer, T. Leijtens, L. M. Herz, A. Petrozza and H. J. Snaith, Science, 2014, 342, 341–344 CrossRef PubMed.
- B. Conings, L. Baeten, C. De Dobbelaere, J. D’Haen, J. Manca and H. G. Boyen, Adv. Mater., 2014, 26, 2041–2046 CrossRef CAS PubMed.
- X. Wei, T. Xie, D. Xu, Q. Zhao, S. Pang and D. Wang, Nanotechnology, 2008, 19, 275707 CrossRef PubMed.
- B. C. O’Regan, K. Bakker, J. Kroeze, H. Smit, P. Sommeling and J. R. Durrant, J. Phys. Chem. B, 2006, 110, 17155–17160 CrossRef PubMed.
- T. Su, Y. Yang, Y. Na, R. Fan, L. Li, L. Wei, B. Yang and W. Cao, ACS Appl. Mater. Interfaces, 2015, 7, 3754–3763 CAS.
- J.-H. Im, I.-H. Jang, N. Pellet, M. Grätzel and N.-G. Park, Nat. Nanotechnol., 2014, 9, 927–932 CrossRef CAS PubMed.
- J. Shi, Y. Luo, H. Wei, J. Luo, J. Dong, S. Lv, J. Xiao, Y. Xu, L. Zhu and X. Xu, ACS Appl. Mater. Interfaces, 2014, 6, 9711–9718 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra12205a |
| This journal is © The Royal Society of Chemistry 2016 |