
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dispersal of pristine graphene for biological studies†
A. P. A.
Raju
a,
S. C.
Offerman
b,
P.
Gorgojo
a,
C.
Vallés
a,
E. V.
Bichenkova
b,
H. S.
Aojula
b,
A.
Vijayraghavan
a,
R. J.
Young
ac,
K. S.
Novoselov
cd,
I. A.
Kinloch
 *ac and
D. J.
Clarke
*b
*ac and
D. J.
Clarke
*b
aSchool of Materials, University of Manchester, Oxford Road, Manchester, M13 9PL, UK. E-mail: ian.kinloch@manchester.ac.uk
bManchester Pharmacy School, University of Manchester, Oxford Road, Manchester, M13 9PL, UK. E-mail: david.clarke@manchester.ac.uk
cNational Graphene Institute, University of Manchester, Oxford Road, Manchester, M13 9PL, UK
dSchool of Physics and Astronomy, University of Manchester, Oxford Road, Manchester, M13 9PL, UK
First published on 18th July 2016
Abstract
Contradictions in the reported biocompatibility of graphene-related materials have been attributed to differences in their preparation. Herein, we address the conflicting behavior of different pristine graphene dispersions through their careful preparation and characterization in aqueous media. Although exfoliated in different media, all graphene dispersions were physically similar and comprised few-layer graphene flakes of 100 to 400 nm mean length with relatively defect-free basal planes. The dispersions were colloidally stable, including in physiological saline and when organic solvents were exchanged with water by dialysis, due to their negative zeta potentials (−28 mV to −60 mV) from interaction with water and the different dispersants. Thus, we have been able to establish the influence of pre-association with different dispersants, including those likely to be encountered during the transport and excretion of graphene in vivo. Shear-forced association with neutral phospholipids was transient, as the lipids desorbed to form liposomes, and left a hemolytic dispersion, whereas other dispersions were not hemolytic. High boiling point solvents widely used to exfoliate graphene are toxic and viewed difficult to remove, but were readily removed by simple dialysis. Human serum albumin readily and stably adsorbed in predominantly monomeric form to pristine graphene in physiological saline, which may be expected in vivo. This work shows the large influence that different adsorbates can have on the behaviour of otherwise physically-similar graphene.
Introduction
Graphene-related materials (GRMs) have been proposed for use in biosensors, tissue engineering and drug delivery due to their unique properties,1–3 reported safety4,5 and biocompatibility.6–9 The wide range of different GRMs has, however, complicated research and biocompatibility studies,9 attributed to differences in the number of graphene layers, their size, stiffness, hydrophobicity, surface functionalities, purity and dosage of the material.4–10 Biological investigations with carefully-prepared and characterized graphene materials are thus urged.9,10 Graphene oxide (GO) materials are widely studied because of their attractive chemistries and aqueous stability,7–11 however such properties are achieved at the cost of the electronic properties of pristine graphene.8–14 GO is prepared by the strong oxidation of graphite, resulting in holes at defects in the GO basal plane, and the absorbance of fragments reminiscent of polyaromatic hydrocarbons.15 Pristine graphene, without such covalent modification and associated defects in its basal plane, is much less explored due to perceived difficulties with its dispersion in aqueous media.11,16GRMs are widely reported to stimulate reactive oxygen species (ROS) on uptake into cells, resulting in lipid and DNA damage, triggering cell death.9 Hydrophilic oxidised and smaller-sized GRMs remain well dispersed and may penetrate cell membranes directly,9 but with lower toxicity than less oxidised GRMs.17 However, defect-free graphene materials exfoliated without acidic and oxidising treatments have been widely viewed to be hydrophobic and to produce unstable aqueous dispersions,7–14 when direct penetration of cell membranes may be hindered by repulsion or the larger size of graphene agglomerates in aqueous media.9 Biological consideration has then largely concerned surfactant-dispersed graphene materials. Surfactant-dispersed few-layer graphene (FLG) caused no detectable changes in human lung epithelial cells18 and surfactant polymer-dispersed FLG also caused little lung inflammation.19 However, similarly surfactant-dispersed FLG has been found to affect the metabolic activity of macrophages in a dose-dependent manner, which was associated with the loss of the mitochondrial membrane potential and raised levels of ROS, triggering cell death by apoptosis.20 Lesser concentrations stimulated pro-inflammatory cytokines, but avoided over-activation.21 Larger micron-sized graphene platelets deposited in lung alveoli, where interaction with lung surfactant lipids is possible, with initial indications of possible inflammation risk and production of ROS on uptake by alveolar macrophages.22 However, minimal oxidation and lung inflammation damage was reported on extended studies.23
Recent studies suggest, however, that largely defect-free graphene interacts with water and is not so hydrophobic as to require surfactants to be dispersed in water, which has been attributed to enhanced edge effects with smaller flake sizes.24 Further, like a cleaned graphite surface with a lowered water contact angle, graphene may present as mildly hydrophilic with charge redistribution from adsorbed hydroxyl ions. This has allowed surfactant-free aqueous graphene dispersions to be prepared in weakly basic aqueous solutions with elevated levels of hydroxyl ions.25 Similarly, single layer graphene, with low levels of edge oxidation from air exposure, also remained dispersed in degassed water, attributed to small attractive dispersive forces between graphene sheets and the electrostatic repulsion from adsorbed hydroxyl ions.26 Although physiological electrolytes may screen this electrostatic repulsion25 and promote agglomeration of graphene, the adsorption of biomolecules from physiological media by graphene is, however, also likely to affect its dispersion-agglomeration behaviour and may result in differences in biological activity.27
Herein we consider whether the conflicting behaviour associated with different graphene preparations can be eliminated by careful preparation and characterization of aqueous dispersions of physically-similar few layer graphene. Given the physical similarity of the preparations, we are able to consider for the first time the influence of different surface adsorbates, particularly those likely to be encountered from the first exposure and transport of pristine graphene in vivo. The adsorbates studied include: (i) water with only residual organic solvents used to exfoliate graphene; (ii) bile acids, which are a gut dispersant and major excretory route of non-metabolized aromatic compounds; (iii) serum albumin, which is the major carrier of aromatic compounds in blood; and (iv) phospholipids, which are a major component of lung surfactant dispersal and biomembranes.
Experimental details
Aqueous graphene exfoliated in organic solvents
Graphene dispersions were produced by the direct exfoliation of graphite (500 mg, natural graphite flakes, grade 2369 from Branwell Graphite Ltd. UK, sieved 500 mesh: average lateral dimension ∼200 μm, (ID/IG)graphite ∼ 0.08) in 100 ml DMF (Sigma Aldrich, D158550, ≥99%) or NMP (Sigma Aldrich, M79603, 99%) in a 100 ml flat bottomed reagent bottle (Duran®) positioned in the middle of a bath sonicator (Elmasonic P, 32 W, 37 kHz, 30% power) ∼50 mm above the transducers for 60 h, and held at a temperature between 22 and 26 °C using a cooling coil. The dispersion produced was centrifuged at 4025 × g for 20 min, after which the supernatant was further centrifuged at 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 180 × g for 20 min at room temperature, when the pellets were resuspended in 10–15 ml of fresh solvent to achieve higher concentrations of graphene.28 Dialysis tubing (Sigma D0405, cellulose-based MWCO 12400 Da) was prepared by boiling in aqueous 1 mM ethylene diamine tetra acetic acid (Sigma E9884) solution for 10 min, followed by rinsing in deionized (DI) water. It was further boiled for 10 min in DI water and washed with copious volumes of DI water before use. Solvent dispersions were introduced into dialysis bags and dialyzed against 3 successive 10 fold volumes of DI water, each for 12 h at room temperature, assisted by gentle magnetic stirring. Graphene concentrations were estimated by optical absorption (Fig. S1†) at 660 nm (4632 ml mg−1 m−1 absorption coefficient,25 estimated from >5 different preparations using gravimetric analysis) and were diluted 30–40% by dialysis (e.g., those used were ∼0.33 mg ml−1 G/DMF and ∼0.16 mg ml−1 G/NMP).
180 × g for 20 min at room temperature, when the pellets were resuspended in 10–15 ml of fresh solvent to achieve higher concentrations of graphene.28 Dialysis tubing (Sigma D0405, cellulose-based MWCO 12400 Da) was prepared by boiling in aqueous 1 mM ethylene diamine tetra acetic acid (Sigma E9884) solution for 10 min, followed by rinsing in deionized (DI) water. It was further boiled for 10 min in DI water and washed with copious volumes of DI water before use. Solvent dispersions were introduced into dialysis bags and dialyzed against 3 successive 10 fold volumes of DI water, each for 12 h at room temperature, assisted by gentle magnetic stirring. Graphene concentrations were estimated by optical absorption (Fig. S1†) at 660 nm (4632 ml mg−1 m−1 absorption coefficient,25 estimated from >5 different preparations using gravimetric analysis) and were diluted 30–40% by dialysis (e.g., those used were ∼0.33 mg ml−1 G/DMF and ∼0.16 mg ml−1 G/NMP).
Exfoliation of graphene in aqueous media
Phospholipids (50 mg Egg Lecithin, Lipid Products, UK) in chloroform–methanol (1:1) were mixed with cholesterol (13.8 mg, Sigma Aldrich, C8667) in a 2:1 molar ratio and dried under nitrogen gas flow at room temperature into a film (lipid). Graphite (250 mg) was added while hydrating the lipid film in 50 ml of autoclaved PBS, comprising sodium phosphate-buffered (10 mM pH 7.4) saline (140 mM NaCl). Alternatively, graphene dispersions were produced by direct exfoliation of graphite (5 mg ml−1) in aqueous solutions of 5 mg ml−1 sodium taurodeoxycholate (TDOC, Sigma Aldrich, T0557) or human serum albumin (HSA, Sigma Aldrich, A9511) in PBS buffer. Flat-bottomed reagent bottles (Duran® 100 ml) were positioned ∼50 mm above the transducers in the middle of a bath sonicator (Elmasonic P, 32 W, 37 kHz, 30% power) and held at a temperature between 22 and 26 °C using a cooling coil for an optimized sonication period (36 h for G/TDOC and G/lipid, 24 h for G/HSA). The dispersions produced were centrifuged for 20 min (11180 × g for G/TDOC, 268 × g for G/HSA and G/lipid) to remove unexfoliated graphite. G/HSA and G/lipid dispersions were stored at 4 °C, and briefly sonicated for 5 min before use, while G/TDOC dispersions were stored at room temperature. Graphene concentrations were estimated by optical absorption at 660 nm using 1825 ml mg−1 m−1 absorption coefficient estimated by gravimetric analysis of >5 preparations (e.g., those used were ∼0.1 mg ml−1 G/TDOC, Fig. S1†).28 A washing procedure was carried out to remove excess lipids and HSA from G/lipid and G/HSA preparation. The dispersions were washed at 6708 × g for 10 min followed by resuspending the sediment in fresh PBS buffer by 2 min sonication. This procedure was repeated three times to obtain G/lipid washed and G/HSA washed dispersions.
Nuclear magnetic resonance (NMR) spectrometry
1H NMR spectra were recorded using a Bruker Avance II + spectrometer operating at proton frequencies of 400 MHz. Spectra were acquired and processed using Bruker software Topspin 2.1. NMR data were collected using 128 transients into 65k data points over a spectral width of 8 kHz, with a relaxation delay of 2.4 s between scans. Processing involved multiplication by an exponential window function prior to Fourier transform and phase correction. 1H chemical shifts (δ) are reported in parts per million (ppm) with peak positions measured relative to trimethyl phosphate (TMP), which was used as an internal standard (doublet; centered at 3.8 ppm).Graphene dispersions were dialyzed against 3 successive 10 fold volumes of heavy water (D2O), each for 12 h at room temperature, assisted by gentle magnetic stirring following the same procedure used for dialysis against water (Fig. S3†). TMP (0.099 M) was added as an internal reference to allow estimation of the residual organic solvent content by integration of the 1H NMR signals relative to those generated by 9 equivalent TMP protons from –O–(CH3)3 groups (Fig. S4†).
UV-visible and fluorescence spectroscopy
Graphene preparations were diluted up to 10–25 fold in their respective stock solutions before measurement of UV-vis absorption (Perkin Elmer Lambda 25 and Varian Cary 5000) and fluorescence (Varian Cary Eclipse) in optical grade quartz cuvettes. Lambert–Beer's law was used to obtain the concentration using optical absorbance at 660 nm (using absorption coefficients of 4632 ml mg−1 m−1 for G/DMF and G/NMP, G/lipid and G/HSA, and 1825 ml mg−1 m−1 for the smaller G/TDOC flakes). Variations in size and thickness may result in differences in absorption coefficients (Fig. S1†).29Atomic force microscopy
AFM was used to measure the lateral dimensions and thickness of graphene flakes. Measurements were taken with a Bruker Dimension FastScan using a FastScan-A (Bruker) probe in tapping mode. Samples were prepared either by drop casting or spray coating on freshly-cleaved mica sheets (Agar Scientific, AGG250-2). G/DMF, G/NMP and G/TDOC samples were prepared by spraying (at 1.5 bar nitrogen gas, 50–100 mm distance) ∼0.5 ml of 10 fold diluted dispersions onto mica sheets using an Evolution Solo Airbrush (Harder & Steenbeck). The mica sheets were heated up to 100 °C prior to spraying, as it helped flash evaporate the solvent during the spraying procedure and reduced the probability of graphene aggregation. The mica sheets were heated on a hot plate at 150 °C to remove excess solvents. This procedure was generally repeated 3–5 times for good coverage. Residual solvents were removed by annealing the substrates under argon atmosphere at 400 °C for 4 h prior to AFM measurements. G/lipid and G/HSA (50 μl of 10 fold diluted dispersions) were drop casted onto freshly-cleaved mica followed by overnight drying at room temperature. Samples were washed with DI water to remove excess lipids and proteins, and gently dried using nitrogen gas.Raman spectroscopy
Graphene laminates were prepared by vacuum filtering the G/DMF, G/NMP (undialyzed and dialyzed) and G/TDOC dispersions obtained after centrifugation onto porous aluminium oxide filter membranes (Whatman® Anodisc, 100 nm, 47 mm diameter) followed by drying in a tube furnace at 400 °C under an argon atmosphere for 4 h. The laminates were analyzed using a Renishaw 2000 spectrometer equipped with 633 nm HeNe laser (1.96 eV, <1 mW). G/lipid and G/HSA Raman spectra were obtained similarly to AFM samples by drop casting samples onto mica substrates.Hydrodynamic size and zeta potential
The hydrodynamic sizes of the graphene dispersions were estimated using a Zetasizer Nano ZS (Malvern Instruments, UK) with undiluted samples (∼1.5 ml) in quartz cuvettes (Fisher Scientific, 12731685). Zeta potential values were also estimated using the same Zetasizer by diluting graphene dispersions 10–25 fold into disposable capillary cells (Malvern DTS1061). All hydrodynamic size and zeta potential estimates are the means of 3–5 measurements.Results and discussion
Water dispersion of solvent-exfoliated graphene
Direct liquid-phase exfoliation (LPE) of graphite to produce pristine FLG at high concentrations is well established using sonication in organic solvents (NMP & DMF) with similar surface tensions to graphene.11–13,29 Although these water-miscible solvents are cytotoxic, particularly NMP, cells survive at low solvent levels (e.g., >90% metabolic activity retained at 0.1% NMP; Fig. S2†). Diluting the solvent to these acceptable levels by simple dilution into aqueous media, also reduces the concentration of the graphene by a similar degree, preventing biologically-meaningful concentrations from being achieved. Furthermore, the high boiling points of the organic solvents prevent their distillation to concentrate graphene preparations or to remove solvents. Thus we pursued gentle but extensive dilution (∼1000×) of the solvents by dialysis against water. This dialysis approach retained 30–40% of the starting graphene concentrations (dialyzed [G/DMF] ∼ 0.33 mg ml−1 & [G/NMP] ∼ 0.16 mg ml−1; Fig. S3†). Low levels of residual solvents (<0.1%) were confirmed by integration of 1H-NMR solvent signals (Table 1; Fig. S4†).| FLG dispersion | Hydro-dynamic diameter (DLS) nm | Mean flake length (AFM) nm | Mean number of layers (AFM) | Zeta potential mV | Solvent (1H-NMR)% (v/v) |
|---|---|---|---|---|---|
| G/DMF | ∼170 | 140 ± 70 | 4.5 ± 2.3 | −48.9 (pH ∼7.6) | 0.07 ± 0.01 |
| G/NMP | ∼200 | 170 ± 80 | 4.3 ± 1.9 | −47.7 (pH ∼ 7.7) | 0.07 ± 0.01 |
| G/TDOC | ∼100 | 90 ± 40 | 3.2 ± 1.8 | −63.0 (pH ∼ 7.6) | NA |
| G/lipid | ∼350 | 350 ± 150 | 5.3 ± 2.1 | ND | NA |
| G/HSA | ∼390 | 370 ± 120 | 6.1 ± 3.0 | −27.9 (pH ∼ 6.8) | NA |
The aqueous graphene dispersions were characterized as being physically similar to the starting FLG dispersed in organic solvents. Atomic force microscopy (AFM) confirmed similar mean flake lengths (<200 nm) and few (<5) graphene layers per flake, indicating little aggregation (Table 1 and Fig. S5†).
Raman bands remained typical of solvent-exfoliated graphene with <5 layers (Fig. S6†). As the solvent exfoliations were performed by well established procedures, the level of edge defects and oxidation from air is expected to be similar to well-reported levels11–14 and further characterisation by XPS was not performed. The lack of broadening in the Raman D and G band suggests that the basal plane had not suffered any severe basal plane disruptions as seen in GO.30
Colloidal solutions with significant zeta potentials (ζ > ±30 mV) are normally considered to be stable. Similar negative values (ζ ∼ −30 mV) have been calculated for the charge transfer from organic solvent to graphene, contributing to the colloid stability of FLG in organic solvents.31 The higher values (ζ > −45 mV, Table 1) for the aqueous FLG measured here suggest that electrostatic repulsion between the FLG sheets similarly contributed to the colloidal stability of these aqueous dispersions. Additional to charge transfer from residual solvent associated with the FLG surfaces,31 water predominates in these dispersions and may be responsible for the higher surface charge.24–26,32 Enhanced edge effects, greater for smaller graphene flakes may also have contributed due to non-covalent bonding between edge carbon atoms and oxygen in water.24 The dispersions were stable at room temperature with slow aggregation leading to a gravitational sedimentation of <25% of the FLG after 10 days and 55–60% after a month, similar to the FLG dispersions in organic solvents (Fig. 1).
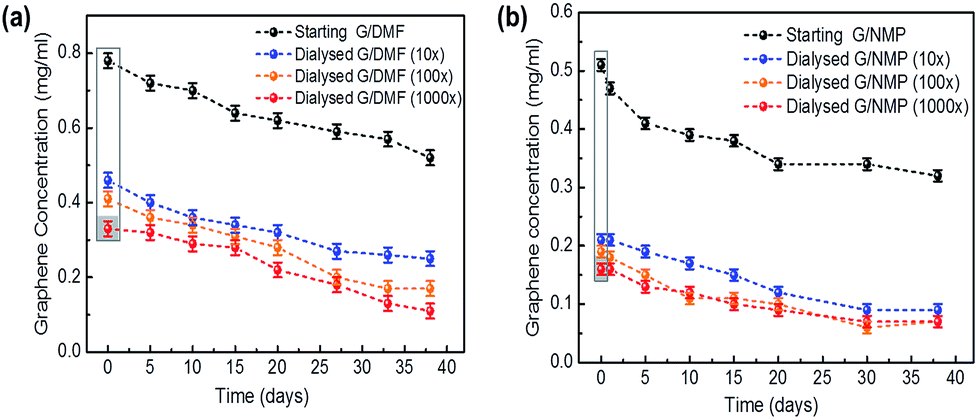 | ||
| Fig. 1 Colloidal stability of dialysed graphene. The sedimentation behaviors of graphene dispersions dialyzed into deionized water (blue trace, >10× dilution; orange trace, >100× dilution; red trace, >1000× dilution of solvent) were compared with the starting graphene dispersed in organic solvents (upper black traces): (a) DMF (left, G/DMF) and (b) NMP (right, G/NMP). Unsedimented graphene concentrations remaining dispersed in colloidal solution, after standing undisturbed at room temperature for over a month (38 days) were estimated by their optical absorbance at 660 nm (4632 ml mg−1 m−1 absorption coefficient). The grey box indicates dilution resulting largely from the expansion of the dialysis bags. | ||
Preparations were re-dispersed by brief sonication for 5 min before use, as also typical for solvent-dispersed FLG. The water-dispersed graphene preparations with solvents diluted by more than 1000 fold showed no significant hemolytic activity at concentrations up to 100 μg ml−1 (Fig. S7†).
Bile surfactant-exfoliated graphene
Bile surfactants provide an excretion route for non-metabolized hydrophobic compounds in the body. Sodium taurodeoxycholate (TDOC) has been used for LPE of FLG, with electrostatic repulsion from its high zeta potential (ζ > −60 mV) providing colloidal stability, but with basal plane defects where intense or lengthy sonication was used.33,34Herein aqueous graphene dispersions were produced by LPE of graphite in aqueous TDOC solution (5 mg ml−1) above its critical micelle concentration, when sonication for up to 36 h to avoid basal plane defects achieved sufficient graphene with high zeta potential (Table 1). Raman spectra (Fig. S8c†) were similar to FLG exfoliated in organic solvents. The 2D band was broader and downshifted compared to the starting graphite, with the ratio of the intensities of the 2D and G peaks (Fig. S8c†), I2D/IG ∼ 0.75, suggesting few-layer graphene (<5 layers).35–37 The characteristic G band appearing around ∼1580 cm−1, with an intense D band (∼1330 cm−1) and D′ band (∼1617 cm−1) indicated the possible presence of defects. However, this was more likely due to the greater influence of the edges of these relatively small flakes (∼100 nm by DLS in Fig. S8d† and ∼90 nm by AFM in Fig. S8e†). The lack of G and D′ band broadening by such defects (these bands merge with high disorder) and the ratio of D to D′ band (ID/ID′ ∼ 4.15) also suggest edge boundary rather than basal plane defects.35–37 High levels of TDOC are already known to remain associated with graphene,33,34 when XPS analysis would reflect the presence of TDOC, but graphene edge oxidation levels.
Phospholipid-exfoliated graphene
GO remains stable in water but, in a physiological saline, the screening of surface electrostatic charge results in GO aggregation.38 Negatively-charged liposomes deposited as lipid monolayers around reduced GO (rGO), enhancing its colloidal stability by electrostatic repulsion. However, neutral liposomes failed to stabilize aqueous rGO, and removal of excess positively-charged liposomes caused rGO agglomeration.39,40Pristine FLG has been directly exfoliated from graphite in the neutral lipid phosphatidylcholine (PC) in chloroform by formation of reverse micelles on the graphene surface.41 FLG is also reported to be exfoliated into liposomes with stable location between the hydrocarbon chains of the phospholipid bilayer.42 Here graphene was directly exfoliated from graphite, by low power sonication for ∼36 h to avoid basal plane defects, by adding it to a neutral lipid film (2:1 molar ratio of egg PC and cholesterol) hydrating in phosphate buffered saline (PBS, 140 mM NaCl pH 7.4). Following removal of unexfoliated graphite and larger liposomes by centrifugation, lipid-exfoliated graphene dispersions (G/lipid) were harvested (Fig. 2) and found to be colloidally-stable (Fig. S9†). Though aggregation was observed after 7 days, dispersion stability was readily regained with shaking and 5 min sonication. This suggests that the neutral lipids associated with pristine graphene and avoided aggregation in saline for longer than for rGO dispersions.39,40
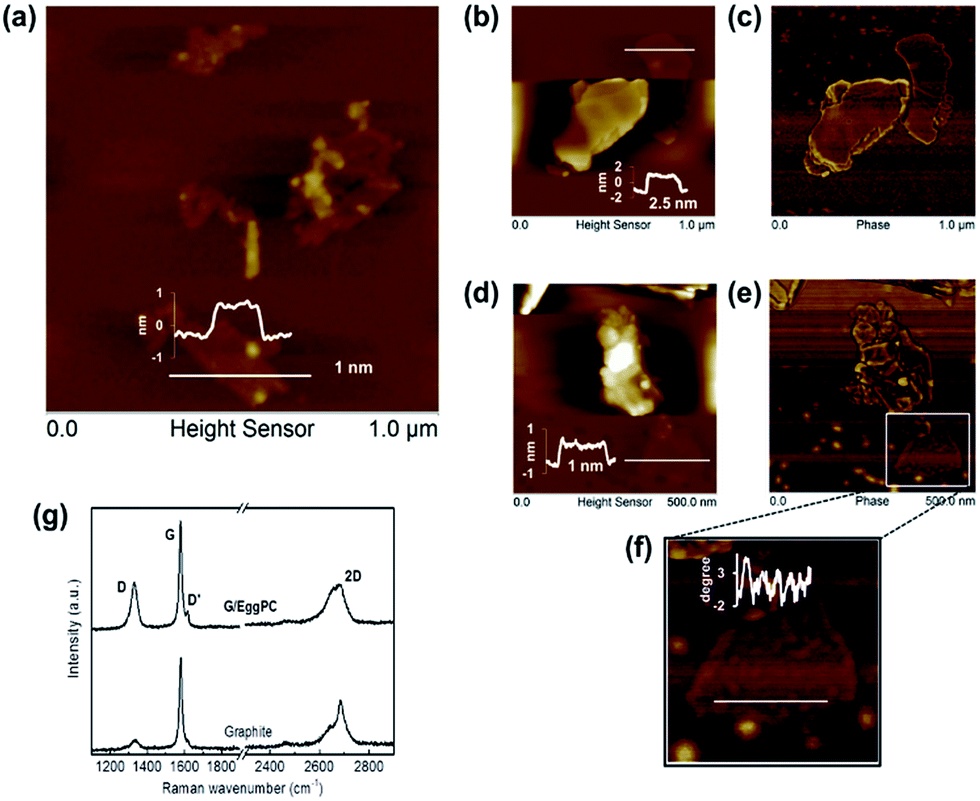 | ||
| Fig. 2 Egg phosphatidylcholine/cholesterol-exfoliated graphene (G/lipid) characteristics. AFM images after drop-casting onto mica and rinsing with deionized water: (a) representative image with a typical inset height scan used to estimate the average flake length (〈L〉 ∼ 350 nm) and the average number of graphene layers (〈N〉 ∼ 5.3); (b) & (d) selected height images of graphene flakes with inset typical height scans; (c) and (e) corresponding phase images (of (b) & (d) respectively). (f) Enlarged AFM phase image of region highlighted in (e) with inset scan showing residual adsorbed lipid molecules. (g) Raman spectra of G/lipid (drop casted on mica substrates) showing few-layer graphene (upper trace) and starting graphite for reference (lower trace). Intensity ratios of the labelled Raman bands were: I2D/IG ∼ 0.45 ± 0.01, ID/IG ∼ 0.31 ± 0.3 and ID/ID ∼ 3.62 ± 0.3. | ||
G/lipid was indicated as few-layer graphene by AFM (Table 1 and Fig. 2) and by the typical shape of the Raman 2D band (Fig. 2g), with I2D/IG ∼ 0.45.34 The presence of the D′ band, with ID/ID′ ∼ 3.52, with no obvious broadening of the D and G bands, suggests that the basal planes of the graphene were relatively defect free, and the D band with ID/IG ∼ 0.31 was mainly due to edge defects.36
Diluted G/lipid dispersions showed two clear size distributions by DLS with average hydrodynamic diameters of ∼60 nm of similar size to sonicated lipid without graphene and ∼400 nm attributed to the lipid-coated graphene, which was a little smaller (∼350 nm) on washing (Fig. S9†). This hydrodynamic diameter agreed with the average flake length determined by AFM (Table 1). The size estimated by the AFM flake length and hydrodynamic diameter of the FLG here appears to be too large to become embedded between the bilayer of the liposomes.42
However, no adsorbed lipid layers were seen on the graphene surface by AFM after washing (Fig. 2), although some residual lipid molecules were seen on thin graphene surfaces (Fig. 2f, phase degree difference43), suggesting a weak interaction between graphene and lipids, which was unstable and readily washed off.
Such weak interactions may also explain the dynamics of aggregation of lipidic FLG (G/lipid), as the weakly-adsorbed neutral lipids leave graphene to assemble into more thermodynamically-stable states in aqueous media, typically liposomes. When G/lipid was extruded under high pressure through 400 nm polycarbonate membrane filters, removing larger FLG flakes, the shear forces dispersed most of the lipid and FLG into a broad size distribution. However, after 10 min, the size distribution split and, after 30 min, two different sizes emerged, which were attributed to lipid (50–100 nm, Fig. S9†) and graphene–lipid aggregates ∼350 nm (Fig. 3). Sonication of this separated dispersion returned a similar broad distribution across both side ranges. Shear forces exerted during extrusion and sonication disrupt liposomes, when their lipids are more available to associate with graphene, but which may not be as thermodynamically stable in aqueous media as lipid–lipid association. In a saline environment, the surface negative charge of graphene would be screened by Na+ cations,25,38 causing some aggregation as the lipids desorbed (Fig. 3).
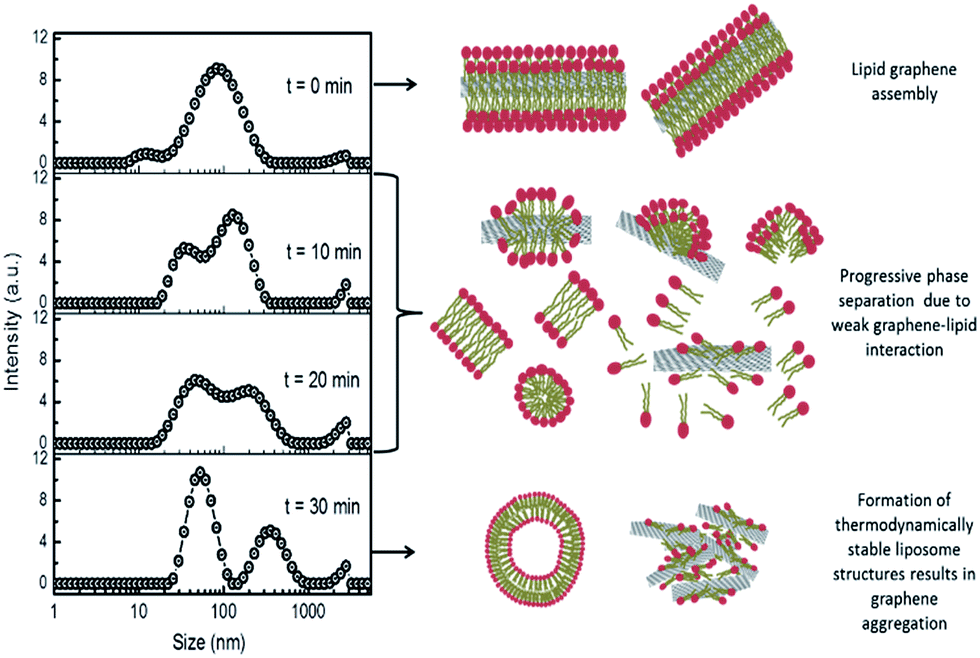 | ||
| Fig. 3 Dynamic change in intensity size distributions (left) following high pressure membrane filtration (400 nm pores) of G/lipid with cartoons (right) of possible transition of unstable phospholipid and graphene assemblies into more thermodynamically-stable structures of liposomes and weakly-agglomerated graphene. | ||
Neutral lipid liposomes adsorbed weakly to graphene, but with spontaneous disruption of those liposomes adsorbed.44 Their stronger adsorption to more oxidised GO and reduced GO, but with slower disruption, suggested that the activation energy for interaction with unoxidised graphene was higher and may involve dispersive extraction of lipids.44 Shear-forced disruption of liposomes by sonication or extrusion may be expected to promote such dispersive extraction of lipids.
Liposomes have been widely used to coat lipid layers onto graphene films, which show a variety of morphologies and structures.39,40 From AFM and modelling studies, the lipid aliphatic chains interact with the aromatic regions of graphene in ordered parallel arrangements, which were relatively sparsely arranged compared to densely-packed liposome membranes.40 Similar arrangements have been suggested for the destructive extraction of lipids from bacterial membranes by graphene sheets.45 Modelling studies suggest that such dispersive extraction of lipids would be stronger for concave hydrophobic graphene surfaces, resulting in hemispherical extracted lipid assemblies. However, in the case of flat or convex surfaces, extracted lipids would be comparatively sparse and tend to move back into lipid membranes.46 Together with studies suggesting that graphene may be mildly hydrophilic24–26 with a negative zeta potential in water (Table 1), findings here appear consistent with the return of excess shear-force extracted lipids back into liposomes.
The penetration of graphene nanosheets into cell membranes is proposed to initiate and propagate along their edge asperities.47–50 Any graphene surrounded by a stable micelle of lipid or surfactant molecules may hinder such edge penetration of cell membranes,48 unless the high curvature of micellar arrangements at the edges are relatively unstable. Edge penetration of graphene covered with a lower density of lipid molecules may not be hindered.48 However, rather than interacting initially by edge penetration, larger FLG sheets may adsorb on the surface of membranes, where lipids may re-orientate and cause greater disruption of the underlying bilayer membrane structure.50 Herein, neutral lipid-exfoliated FLG was hemolytic, disrupting red blood cell membrane to release haemoglobin (G/lipid at >5 μg ml−1, Fig. S7†), and also inhibited cell metabolic activity (Fig. S10†), whereas FLG of similar physical characteristics dispersed in water was not hemolytic and bile surfactant-dispersed FLG was only weakly haemolytic (Fig. S7†). This suggests that loosely-packed lipids remaining on the FLG may have participated in a biomembrane disruption process with FLG. Sparse lipid coating with aliphatic hydrocarbon chains lying on an otherwise mildly hydrophilic graphene surface24–26 may facilitate interaction with the membrane bilayer. Cell studies, in support of the edge penetration of similarly-sized FLG, also dispersed their FLG with neutral lipids (di-palmitoyl phosphatidylcholine) by vortex mixing in aqueous ethanol,49 which may be expected to extract phospholipid molecules onto FLG. However, this FLG was then treated with albumin,49 which may also adsorb onto FLG and influence its interaction with phospholipid bilayers and biomembrane receptors.
Albumin-exfoliated graphene
Human serum albumin (HSA), as the most abundant protein in blood plasma, with conformational flexibility and binding of hydrophobes, is well suited to exfoliating51 and to stabilizing graphene under physiological conditions.52 Graphene could be exfoliated directly from graphite by sonication in HSA (∼5 mg ml−1 and 24 h sonication were optimal). Higher HSA concentrations and lengthier sonication times resulted in protein aggregation and less graphene exfoliation in PBS solutions (Fig. S11†), although graphene exfoliation did not decline in water solutions at higher bovine serum albumin concentrations.51 However, AFM studies observed higher order aggregated network structures formed on graphite surfaces upon adsorption of HSA from higher concentration solutions.52Graphene flakes of average length ∼370 nm had higher overall thickness (7.6 ± 1.5 nm), with individual HSA molecules but no HSA networks or aggregates visible on graphene surfaces by AFM (Fig. 4). The average height of HSA molecules (∼2 nm)52 was adsorbed on both sides of the FLG, when the average number of graphene layers was estimated as 6.1 ± 3.0. The shape of the Raman 2D band and I2D/IG ∼ 0.48 indicates few-layer graphene35–37 agreeing with the AFM analysis. The presence of a D′ band with ID/ID′ ∼ 4.3, with no obvious broadening of D and G band suggests that the basal plane of graphene was relatively defect free,35–37 and the D band with ID/IG ∼ 0.5 was due mainly to edge defects (Fig. S12†).
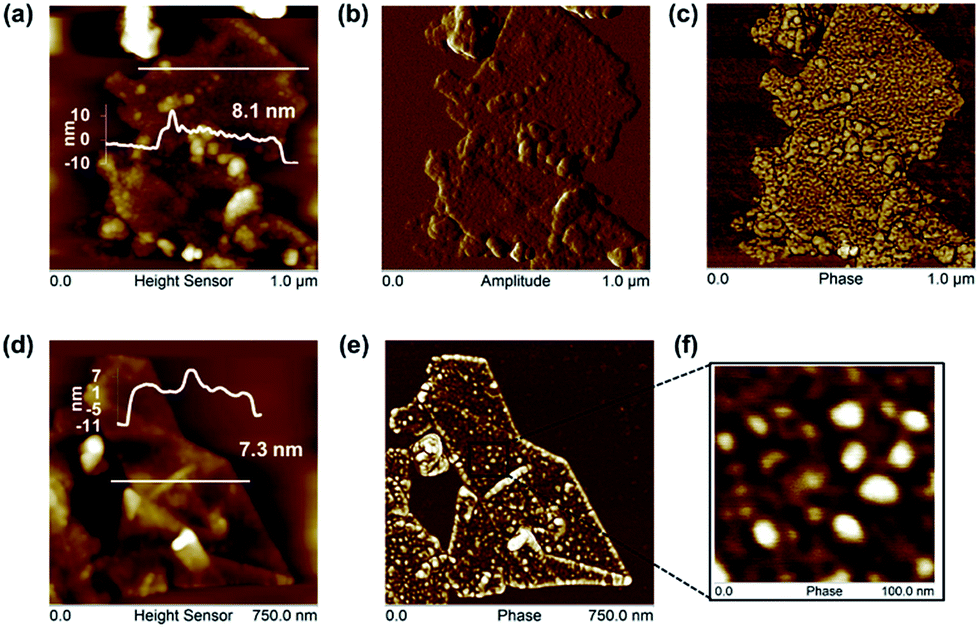 | ||
| Fig. 4 Representative AFM images of HSA-exfoliated graphene (G/HSA) showing strong adsorption of monomeric HSA without aggregation. Top row: (a) height sensor images with typical height scans used to estimate the average observed thickness, (b) corresponding amplitude and (c) phase images of (a) showing the extent of HSA coverage on the FLG surface. Bottom row: further (d) height sensor and (e) corresponding phase images showing clear HSA adsorption on FLG surface and at its edges; and (f) enlarged phase image of indicated region of (e) showing several individual ellipsoidal HSA molecules and no HSA aggregates. | ||
We suggest that the HSA was predominantly adsorbed, rather than chemisorbed, as expected for retention on gold surfaces by Au–S coordination bonding, deriving from the cystine bridges retaining albumin structure.53 There was a lack of evidence of basal plane defects, which would be evident for any extensive covalent attachment of the adsorbed HSA. HSA adsorbed predominantly across the surface of the FLG flakes, with no greater levels at the edges. Edge defects did not appear to be a site of preferential adsorption, where any covalent retention might be expected.
The adsorption of albumin and other proteins to graphite and graphene surfaces has been suggested to result in unfolding and denaturation, whereas other theoretical studies with explicit description of solvent (water) suggest the opposite, and the preservation of binding sites in the most studied serum bovine albumin.54 Predominantly monomeric albumin molecules were observed on FLG surfaces by AFM with no denatured aggregates or networks (Fig. 4).
With an isoelectric point of ∼4.7, at physiological pH,52 the anionic charge density due to HSA carboxylate ions would be expected to dominate the surface of the G/HSA adsorbate. Though the resulting electrostatic repulsion provided a stable dispersion, the zeta potential (ζ ∼ −27.9 mV) fell below levels effective for colloidal stability upon washing in saline (ζ ∼ −13.4 mV), when slow aggregation would be promoted by the increased electrostatic screening by Na+ cations.25,38,51 Removal of unadsorbed HSA and resuspension in PBS without added HSA also reduced colloidal stability (Fig. S11†), presumably from loss of adsorbed HSA back into solution.
HSA interaction with FLG was followed by hydrodynamic sizing (DLS) and by tryptophan fluorescence quenching (Fig. 5). Upon addition of dialyzed, water-dispersed G/DMF to HSA solutions in PBS, a progressive shift in size distribution was clear (Fig. 5a), as HSA monomers (∼3.8 nm) associated with FLG (∼100 nm), resulting in the growth of a population of slightly larger hydrodynamic size, presumably graphene–HSA adsorbates (Fig. 5a). HSA could no longer be detected in the presence of excess graphene, which resulted in a broader size distribution of graphene–HSA.
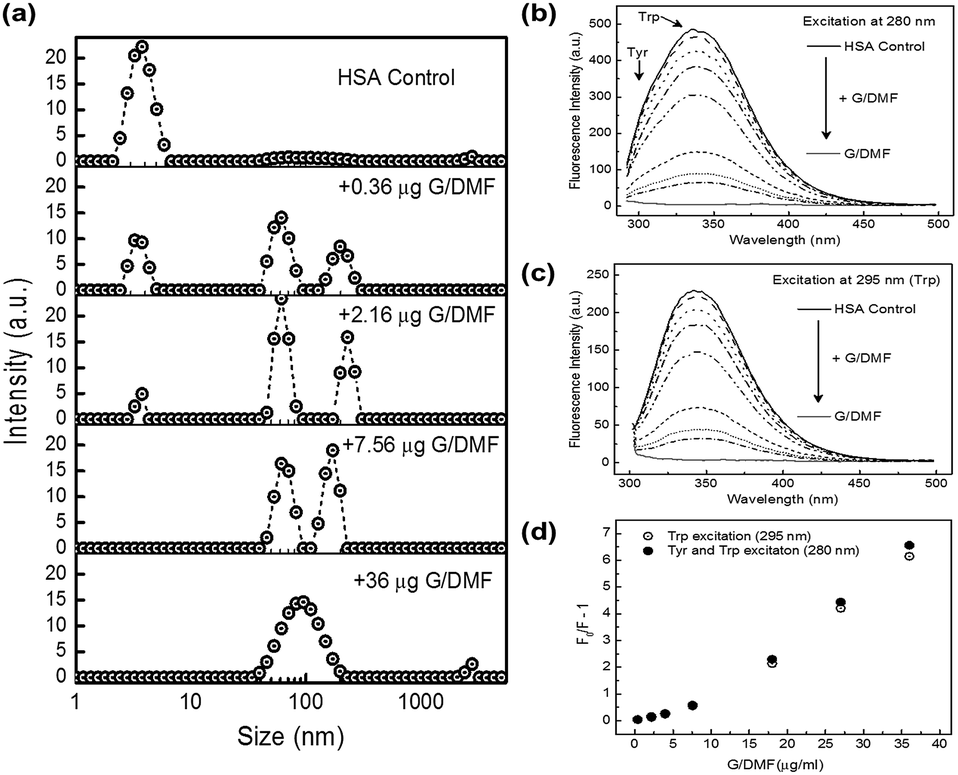 | ||
| Fig. 5 Albumin binding to graphene followed by change in size and fluorescence. (a) Change in hydrodynamic size distribution by DLS on addition of increasing concentrations of dialyzed few layer graphene (G/DMF) to human serum albumin (HSA, 5 mg ml−1 in PBS buffer, filtered through 20 nm Whatman® Anodisc filters). Quenching of fluorescence emission of HSA on addition of similar increasing concentrations of graphene upon excitation (b) at 280 nm and (c) at 295 nm, where traces from top to bottom are HSA (5 mg ml−1) alone and upon sequential addition of 0.4, 2.2, 4, 7.6, 18, 27 and 36 μg G/DMF to the HSA, and dialyzed G/DMF alone. (d) Stern–Volmer plot of quenching of fluorescence emission by G/DMF upon excitation at 280 nm (closed circles) and 295 nm (open circles). | ||
Tryptophan fluorescence was quenched by association with the same concentrations of FLG (Fig. 5b and c). The Stern–Volmer plot (Fig. 5d) indicated predominantly static quenching (tryptophan involved in HSA adsorption to graphene) with a contribution from dynamic quenching.55 XPS studies also infer albumin adsorption on graphene surfaces51 and so was not considered here to inform on the oxidation state of edge defects.
FLG is expected to aggregate in PBS,38–40 which aggregation has been suggested to reduce its hemolytic activity.56 However, FLG freshly exfoliated from graphite suspended in PBS supplemented with HSA remained well dispersed with adsorbed monomeric HSA, without significant haemolytic activity, as found for water and bile surfactant dispersed FLG (Fig. S7†) nor effect on cell activity of breast cancer and mouse macrophage cells (Fig. S10†). Water-dispersed graphene introduced into HSA solutions remained well dispersed in PBS by binding of HSA, removing HSA to levels undetectable by DLS and almost fully quenching its tryptophan fluorescence through binding to graphene (Fig. 5). Similar HSA dispersal of graphene would also be expected in blood serum. The underlying adsorption of HSA may also be expected to block any hemolytic activity,53,56 and progressively replace other loosely associated adsorbates including phospholipids. Albumin-modified nanoparticles tend to be taken into cells of the reticulo-endothelial system by FcRn receptor-mediated endocytosis, and recycled back into circulation.57
Conclusions
Aqueous exfoliation of graphene from graphite by sonication and adsorption of very different biomolecules (HSA, bile salt & phospholipids) produced pristine graphene with few layers (average < 5 layers) and size (100–400 nm) in the range of physical characteristics typical of the well-established exfoliation of FLG in organic solvents (NMP & DMF). Aqueous dispersions remained stable with negative zeta potentials (−20 mV to −60 mV) at neutral pH sufficient for electrostatic repulsion. Given the relative similarity of the physical characteristics of the dispersions, hemolytic activity (∼30% at 50 μg ml−1 graphene) was attributed to weak association with phospholipid molecules.Conflict of interests
The authors declare no competing financial interests.Author contributions
A. P. A. R. prepared and characterized graphene and analyzed the data under the supervision of I. A. K., R. J. Y., D. J. C., and the direction of K. S. N., S. C. O. performed the cell studies and analyzed the data under the supervision of H. S. A. and D. J. C., A. P. A. R. and P. G. carried out the AFM analysis. E. V. B. and A. P. A. R. performed NMR and analyzed the data, A. P. A. R. and D. J. C. co-wrote the paper and contributed to methodology development. All authors discussed the results and commented on the manuscript.Acknowledgements
A. P. A. R. is grateful for the support of the Dean's Scholarship and the Graphene Environment, Health & Safety Committee, University of Manchester. C. V., R. J. Y. and I. A. K. acknowledge the EU Graphene Flagship grant agreement no. 604391 and the EPSRC (EP/I023879/1). The authors acknowledge financial support from the Graphene Bioscience Interdisciplinary Grand Challenges, University of Manchester, UK.References
- K. V. Krishna, C. Ménard-Moyon, S. Verma and A. Bianco, Nanomedicine, 2013, 8, 1669–1688 CrossRef CAS PubMed.
- K. Kostarelos and K. S. Novoselov, Nat. Nanotechnol., 2014, 9, 744–745 CrossRef CAS PubMed.
- J. Liu, L. Cui and D. Losic, Acta Biomater., 2013, 9, 9243–9257 CrossRef CAS PubMed.
- V. C. Sanchez, A. Jachak, R. H. Hurt and A. B. Kane, Chem. Res. Toxicol., 2012, 25, 15–34 CrossRef CAS PubMed.
- C. Bussy, H. Ali-Boucetta and K. Kostarelos, Acc. Chem. Res., 2013, 46, 692–701 CrossRef CAS PubMed.
- A. M. Jastrzębska, P. Kurtycz and A. R. Olszyna, J. Nanopart. Res., 2012, 14, 1320–1328 CrossRef PubMed.
- A. Bianco, Angew. Chem., Int. Ed., 2013, 52, 4986–4997 CrossRef CAS PubMed.
- A. B. Seabra, A. J. Paula, R. de Lima, O. L. Alves and N. Durán, Chem. Res. Toxicol., 2014, 27, 159–168 CrossRef CAS PubMed.
- A. M. Pinto, I. C. Gonçalves and F. D. Magalhães, Colloids Surf., B, 2013, 188–202 CrossRef CAS PubMed.
- K. Kostarelos and K. S. Novoselov, Science, 2014, 344, 261–263 CrossRef CAS PubMed.
- M. Nurunnabi, K. Parvez, M. Nafiujjaman, V. Revuri, H. A. Khan, X. Feng and Y. Lee, RSC Adv., 2015, 5, 42141–42161 RSC.
- D. W. Johnson, B. P. Dobson and K. S. Coleman, Curr. Opin. Colloid Interface Sci., 2015, 20, 367–382 CrossRef CAS.
- A. Ciesielski and P. Samorı, Chem. Soc. Rev., 2014, 43, 381–398 RSC.
- A. Sasidharan, L. S. Panchakarla, P. Chandran, D. Menon, S. Nair, C. N. R. Rao and M. Koyakutty, Nanoscale, 2011, 3, 2461–2464 RSC.
- J. P. Rourke, P. A. Pandey, J. J. Moore, M. Bates, I. A. Kinloch, R. J. Young and N. R. Wilson, Angew. Chem., Int. Ed., 2011, 50, 3173–3177 CrossRef CAS PubMed.
- C. Bussy, D. Jasim, N. Lozano, D. Terry and K. Kostarelos, Nanoscale, 2015, 7, 6432–6435 RSC.
- A. M. Pinto, C. Gonçalves, D. M. Sousa, A. R. Ferreira, J. A. Moreira, I. C. Gonçalves and F. D. Magalhães, Carbon, 2016, 99, 318–329 CrossRef CAS.
- J. Conroy, N. K. Verma, R. J. Smith, E. Rezvani, G. S. Duesberg, J. N. Coleman and Y. Volkov, arXiv:1406.2497v1 [q-bio.CB], 2014.
- M. C. Duch, G. R. S. Budinger, Y. T. Liang, S. Soberanes, D. Urich, S. E. Chiarella, L. A. Campochiaro, A. Gonzalez, N. S. Chandel, M. C. Hersam and G. M. Mutlu, Nano Lett., 2011, 11, 5201–5207 CrossRef PubMed.
- Y. Li, Y. Liu, Y. Fu, T. Wei, L. Le Guyader, G. Gao, R. S. Liu, Y. Z. Chang and C. Chen, Biomaterials, 2012, 33, 402–411 CrossRef CAS PubMed.
- H. Zhou, K. Zhao, W. Li, N. Yang, Y. Liu, C. Chen and T. Wei, Biomaterials, 2012, 33, 6933–6942 CrossRef CAS PubMed.
- A. Schinwald, F. A. Murphy, A. Jones, W. MacNee and K. Donaldson, ACS Nano, 2012, 6, 736–746 CrossRef CAS PubMed.
- A. Schinwald, F. Murphy, A. Askounis, V. Koutsos, K. Sefiane and K. Donaldson, Nanotoxicology, 2014, 8, 824–832 CrossRef CAS PubMed.
- M. Yi, Z. Shen, S. Liang, L. Liu, X. Zhanga and S. Maa, Chem. Commun., 2013, 49, 11059–11061 RSC.
- K. B. Ricardo, A. Sendecki and H. Liu, Chem. Commun., 2014, 50, 2751–2754 RSC.
- G. Bepete, E. Anglaret, L. Ortolani, V. Morandi, A. Pénicaud and C. Drummond, arXiv 1603.05421, 2014.
- J. Zhao, Z. Wang, J. C. White and B. Xing, Environ. Sci. Technol., 2014, 48, 9995–10009 CrossRef CAS PubMed.
- R. Su, S. F. Lin, D. Q. Chen and G. H. Chen, J. Phys. Chem. C, 2014, 118, 12520–12525 CAS.
- J. N. Coleman, Acc. Chem. Res., 2013, 46, 14–22 CrossRef CAS PubMed.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS.
- W. W. Liu, J. N. Wang and X. X. Wang, Nanoscale, 2012, 4, 425–428 RSC.
- A. N. Camden, S. A. Barr and R. J. Berry, J. Phys. Chem. B, 2013, 117, 10691–10697 CrossRef CAS PubMed.
- M. Lotya, P. J. King, U. Khan, S. De and J. N. Coleman, ACS Nano, 2010, 4, 3155–3162 CrossRef CAS PubMed.
- Z. Sun, J. Masa, Z. Liu, W. Schuhmann and M. Muhler, Chemistry, 2012, 18, 6972–6978 CrossRef CAS PubMed.
- A. C. Ferrari, J. C. Meyer, V. Scardaci, C. Casiraghi, M. Lazzeri, F. Mauri, S. Piscanec, D. Jiang, K. S. Novoselov, S. Roth and A. K. Geim, Phys. Rev. Lett., 2006, 97, 187401 CrossRef CAS PubMed.
- R. Beams, L. G. Cançado and L. Novotny, J. Phys.: Condens. Matter, 2015, 27, 083002 CrossRef PubMed.
- A. Eckmann, A. Felten, A. Mishchenko, L. Britnell, R. Krupke, K. S. Novoselov and C. Casiraghi, Nano Lett., 2012, 12, 3925–3930 CrossRef CAS PubMed.
- C.-J. Shih, S. Lin, R. Sharma, M. S. Strano and D. Blankschtein, Langmuir, 2012, 28, 235–241 CrossRef CAS PubMed.
- J. Liu, S. Guo, L. Han, T. Wang, W. Hong, Y. Liu and E. Wang, J. Mater. Chem., 2012, 22, 20634–20640 RSC.
- L. Rui, J. Liua, J. Li, Y. Weng, Y. Dou, B. Yuana, K. Yang and Y. Ma, Biochim. Biophys. Acta, 2015, 1848, 1203–1211 CrossRef CAS PubMed.
- M. Pykal, K. Šafářová, K. M. Šišková, P. Jurečka, A. B. Bourlinos, R. Zbořil and M. Otyepka, J. Phys. Chem. C, 2013, 117, 11800–11803 CAS.
- R. Zappacosta, M. Di Giulio, V. Ettorre, D. Bosco, C. Hadad, G. Siani, S. Di Bartolomeo, A. Cataldi, L. Cellini and A. Fontana, J. Mater. Chem. B, 2015, 3, 6520–6527 RSC.
- S. J. Attwood, Y. Choi and Z. Leonenko, Int. J. Mol. Sci., 2013, 14, 3514–3539 CrossRef CAS PubMed.
- A. C.-F. Ip, B. Liu, P.-J. J. Huang and J. Liu, Small, 2013, 9(7), 1030–1035 CrossRef CAS PubMed.
- Y. Tu, M. Lv, P. Xiu, T. Huynh, M. Zhang, M. Castelli, Z. Liu, Q. Huang, C. Fan, H. Fang and R. Zhou, Nat. Nanotechnol., 2013, 8, 594–601 CrossRef CAS PubMed.
- B. Luan, T. Huynh and R. Zhou, Nanoscale, 2016, 8, 5750–5754 RSC.
- X. Shi, A. von dem Bussche, R. H. Hurt, A. B. Kane and H. Gao, Nat. Nanotechnol., 2011, 6, 714–717 CrossRef CAS PubMed.
- J. Wang, Y. Wei, X. Shi and H. Gao, RSC Adv., 2013, 3, 15776–15782 RSC.
- Y. Li, H. Yuan, A. von dem Bussche, M. Creighton, R. H. Hurt, A. B. Kane and H. Gao, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 12295–12300 CrossRef CAS PubMed.
- M. Dallavalle, M. Calvaresi, A. Bottoni, M. Melle-Franco and F. Zerbetto, ACS Appl. Mater. Interfaces, 2015, 7, 4406–4414 CAS.
- S. Ahadian, M. Estili, V. J. Surya, J. Ramón-Azcón, X. Liang, H. Shiku, M. Ramalingam, T. Matsue, Y. Sakka, H. Bae, K. Nakajima, Y. Kawazoec and A. Khademhosseini, Nanoscale, 2015, 7, 6436–6443 RSC.
- X. Peng, H. Fu, R. Liu, L. Zhao, Y. Zu, F. Xu and Z. Liu, Scanning, 2015, 37, 158–164 CrossRef CAS PubMed.
- L. Wang, J. Li, J. Pan, X. Jiang, Y. Ji, Y. Li, Y. Qu, Y. Zhao, X. Wu and C. Chen, J. Am. Chem. Soc., 2013, 135, 17359–17368 CrossRef CAS PubMed.
- J. G. Vilhena, P. Rubio-Pereda, P. Vellosillo, P. A. Serena and R. Pérez, Langmuir, 2016, 32, 1742–1755 CrossRef CAS PubMed.
- S. Li, A. N. Aphale, I. G. Macwan, P. K. Patra, W. G. Gonzalez, J. Miksovska and R. M. Leblanc, ACS Appl. Mater. Interfaces, 2012, 4, 7069–7075 CAS.
- K.-H. Liao, Y.-S. Lin, C. W. Macosko and C. L. Haynes, ACS Appl. Mater. Interfaces, 2011, 3, 2607–2615 CAS.
- T. Kuo, K. Baker, M. Yoshida, S. W. Qiao, V. G. Aveson, W. I. Lencer and R. S. Blumberg, J. Clin. Immunol., 2010, 30, 777–789 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Includes further details of materials characterisation and cell biological studies in Fig. S1 to S12. See DOI: 10.1039/c6ra12195k |
| This journal is © The Royal Society of Chemistry 2016 |