
One-pot syntheses of N-(α-fluorovinyl)azole derivatives from N-(diphenylmethylene)-2,2,2-trifluoroethanamine†
Abstract
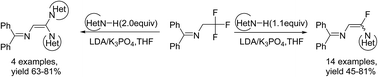
N-(Diphenylmethylene)-2,2,2-trifluoroethanamine acts as a versatile fluorine-containing building block, and from which N-(α-fluorovinyl)azole derivatives were readily prepared in good yields with relatively high stereoselectivity by using various N–H containing heterocycles as nucleophiles via vinylic substitution reactions (SNV) in the presence of LDA and K3PO4 in a one-pot process.
 Please wait while we load your content...
Please wait while we load your content...

