DOI:
10.1039/C6RA12135G
(Paper)
RSC Adv., 2016,
6, 65518-65523
In situ photoacoustic spectroscopic analysis on photocatalytic decolorization of methylene blue over titanium(IV) oxide particles
Received
10th May 2016
, Accepted 5th July 2016
First published on 5th July 2016
Abstract
Photoabsorption of an aqueous suspension containing methylene blue (MB) dye and titanium(IV) oxide (TiO2) powder was evaluated using photoacoustic spectroscopy (PAS). Simultaneous detection of photoabsorption of MB both in solution and adsorbed on TiO2 was performed using a corrosion-resistant cell with microphone detection. In addition, time-course PAS measurements were employed to observe the photocatalytic decolorization of MB in the aqueous suspension. The time-course data of the MB decolorization process attributed to the oxidation or reduction of MB was obtained at 20 s intervals. Moreover, the MB decolorization rate as a function of the initial pH of the suspension was different from that obtained by conventional evaluation methods. This discrepancy most likely stems from the advantages of PAS analysis, which enables the detection of all MB dye included in the aqueous suspension.
1 Introductions
Titanium(IV) oxide (TiO2) particles are representative semiconductor photocatalyst that are used in industry as self-cleaning agents, and employed in coatings to reduce the necessity for cleaning,1,2 and as a novel photocatalyst for environmental remediation has been intensively studied.3–5 One method conventionally used to evaluate the self-cleaning properties of a photocatalyst is measurement of the decolorization of organic dyes, such as methylene blue (MB),6,7 in the presence of the photocatalyst. This method has been standardized by the International Organization for Standardization (ISO).8,9 To conduct this evaluation of a powdered photocatalyst, an aqueous suspension of dye and powder is sampled at regular intervals and centrifuged, and then the resulting supernatant is analyzed by ultraviolet-visible spectroscopy. However, some dye strongly absorbed on the powder cannot be removed from catalytic surface and only “free” dye in the solution other than adsorbed dye are determined colorimetrically. As a result, the photocatalytic activity is determined by the concentration ratio “C/C0”, where C0 and C are the initial and measured concentrations of dye. Thus, photocatalytic activity determined by conventional method depend on adsorption ability of the photocatalyst for the dye, and samples having different adsorption ability for dye, which is mainly determined by physical and chemical properties such as specific surface area, are not evaluated equally.
The pH value is an important factor influencing the photocatalytic reaction rate for suspended metal oxide particles because it affects the adsorption properties of the particles. It is well known that the surface states of metal oxides are influenced by pH. The surfaces of particles become negatively charged for solutions with pH values greater than the point of zero charge (pzc), which is a pH of 5–7 for TiO2 solutions, while pH values lower than the pzc give rise to positively charged surfaces.10–12 In addition, it is believed that solutions with high pH values create favorable environments for degradation of cationic dyes such as MB because of a large electrostatic attraction between the dye and TiO2 surface.13–15 The chemical reaction involved in the degradation of MB can be described as follows.9
| C16H18N3SCl + 25.5O2 → HCl + H2SO4 + 3HNO3 + 16CO2 + 6H2O |
This reaction involves the generation of various acidic species, leading to a subsequent decrease in pH as MB degradation proceeds. This is of consequence since progress of MB degradation changes the adsorption properties of MB on the photocatalyst, and it is difficult to elucidate how pH influences photocatalytic activity of MB degradation. Moreover, these final oxidation products generate through various kinds of intermediate species, and these species possibly affect MB adsorption on the TiO2 surface.
In the present study, photoabsorption of suspensions containing MB dye and TiO2 powder was evaluated using photoacoustic spectroscopy (PAS). In the most of spectroscopic studies, diffuse reflectance spectroscopy has been frequently employed to estimate absorption of powder samples, but sometimes scattering and reflection cause difficulty in accurate measurement of photoabsorption. PAS is one of absorption spectroscopy that is applicable to even opaque and strongly scattering materials because it detects photoabsorption indirectly through photothermal waves generated by the relaxation of the photoexcited states, e.g., recombination of an electron–hole pair in semiconductor and de-excitation of dye molecules.16,17 PAS has already been used to study gas-phase photochemical reactions of dyes adsorbed on semiconductor surfaces,18–20 and we have also utilized it to detect photoabsorption in TiO2 powder under photocatalytic condition.21,22 However, there have been no studies on in situ observations of photocatalytic reaction in liquid suspensions with few exceptions.23,24 In contrast to solid sample analysis, several studies of liquid samples used a high-power pulsed laser as an excitation source and a piezoelectric ceramic (PZT) as an acoustic detector in lieu of a microphone.23–28 The PZT was used because volatile components containing water and acid can negatively affect the microphone and introduce errors. While suitable in liquids, PZT detection is not suitable for measuring suspension samples because light scattering may induce false signals and PZT detection.23 In the present study, we modified a conventional microphone photoacoustic (PA) cell by separating the sample cuvette and microphone chamber with a thin cover glass in order to improve the corrosion resistance. As far as we know, no study on suspension samples using such a corrosion-resistant microphone PA cell has been reported. Herein, we describe the application of a PAS system to observe photocatalytic dye decolorization via changes in photoabsorption in a suspension containing MB dye and TiO2 powder. Photocatalytic reactions under various conditions were observed. Moreover, we determined the dependence of the MB decolorization rate on the initial pH of the suspension using a time-course analysis derived from the PAS measurements.
2 Experimental
2.1. Materials
Analytical grade reagents MB (C16H18N3SCl, Wako Pure Chemical), ethanol, and other chemicals were used without further purification. TiO2 powder supplied by Showa Denko (Super-Titania F6A) was used. The aqueous suspensions were prepared by adding 100 mg of TiO2 to 5 mL of aqueous solution containing typically 50 μmol L−1 of MB. The suspensions were then treated by ultrasound for 5 min and magnetically stirred in the dark for 30 min to ensure that adsorption equilibrium was reached. The pH of the suspension was controlled by adding sulfuric acid and sodium hydroxide. Finally, 1.5 mL of the suspension was transferred into the cuvette of the PA cell. To prepare TiO2 powder modified with MB, 10 mL of an aqueous suspension containing 200 mg of TiO2 and 25 μmol L−1 MB was stirred for 1 h. The unwashed, centrifugally-separated residue was dried at 60 °C under vacuum.
2.2. PA cell
A homemade PA cell with a duralumin body and a quartz window was used. For the measurements on the suspension sample, the PA cell was attached to a UV-transparent disposable cuvette with a 6 mm ϕ hole, which was covered with a cover glass (Matsunami glass, 0.04–0.06 mm), and set up as shown Fig. 1. For powder sample measurements, a powder was packed into the sample holder and placed in the cell. The digital PA signal was acquired by a digital MEMS microphone (Cirrus Logic Inc., WM7211IMSE/RV) buried in the cell, and recorded using a PC equipped with digital I/O interface. Time-series data was acquired by analog conversion of the digital PA signal, followed by a Fourier transform with a Hamming window function.
 |
| | Fig. 1 Schematic illustration of PA cell. | |
2.3. Modulation frequency dependent measurements
Laser diode emitting light at 670 nm (Huanic, MH670-4-5) was used as probe light and its output intensity was modulated by a function generator (Texio, FG281). The sample was illuminated through a window on top of the PA cell. Samples were stirred during measurements under ambient conditions.
2.4. Spectroscopic measurements
Monochromatic light was extracted from the output of a white light emitting diode (Cree, CXA1512-0000-000F0HM240F) using a monochromator (Jasco CT-101T). Spectroscopic measurements were made using light at wavelengths from 400 to 750 nm in 5 nm steps, modulated at 2.5 Hz. The sample was illuminated through a window on top of the PA cell. The measurements were carried out under ambient conditions while the sample was stirred. PA spectra were normalized by that of the reference, carbon black powder, to compensate for the wavenumber dependence of the light intensity.
2.5. Time-course measurements
For the time-course measurements, laser diode emitting light at 670 nm (Huanic, MH670-4-5) was used as probe light and its output intensity was modulated at 2.5 Hz. As with the aforementioned experiments, the illumination pathway to the sample was through the window on top of the PA cell. Ultraviolet (UV) irradiation illuminated the sample through a cuvette, which was opposite side of probe light, using a light-emitting diode (Nichia NCSU033B, emitting around 365 nm, 10 mW cm−2). During measurements, the headspace of the cuvette was purged using nitrogen (N2) or oxygen (O2) gas and the suspension was stirred. The time-course data was obtained at 20 s intervals. Reaction rate constant (k) of MB degradation was calculated by curve fitting of experimental data to a pseudo-first-order model.
3 Results and discussions
3.1. Modulation frequency dependence of PA intensity
Fig. 2 shows the dependence of the PA intensity on the modulation frequency for suspensions containing 20 g L−1 of TiO2 with and without 50 μmol L−1 of MB. The PA intensity detected in the presence of MB was larger than that in the absence of MB, indicating that the PA signal is mainly dependent on photoabsorption of MB. The small increase in PA intensity detected in the absence of MB is due to trace background photoabsorption by the inner wall of the cell. This background signal showed no modulation frequency dependence. In contrast, the PA intensity in the presence of MB increased with decreasing modulation frequency. This increase in intensity is due to an increase in the maximum measurement depth (d) for PAS, which depends on the modulation frequency (f) of the irradiating light source as follows,16
where α is the thermal diffusivity of the medium. Thus, a low modulation frequency is preferable for detecting photothermal waves from a suspension through cover glass. However, the sensitivity and stability of the microphone decreases with decreasing frequency. In practice, the rms error in the PA intensity increases with decreasing modulation frequency. A modulation frequency of 2.5 Hz was found to be optimal for balancing the rms error and signal intensity, and was used in the present study. At f = 2.5 Hz, d for the cover glass is calculated to be ca. 277 μm using α = 6.9 × 10−7 m2 s−1 (borosilicate glass), and a frequency of 2.5 Hz is sufficiently low to detect the PA signal of the suspension. Fig. 3 shows the PA intensity as a function of MB concentration in suspensions containing 20 g L−1 of TiO2. The relationship is seen to be linear, and can therefore be used as a calibration curve for determining unknown MB concentrations based on the measured PA intensity.
 |
| | Fig. 2 Modulation frequency dependence of PA intensity of suspension containing 20 g L−1 of TiO2 with and without 50 μmol L−1 of MB. | |
 |
| | Fig. 3 Dependence of MB concentration on PA intensity in suspensions containing 20 g L−1 of TiO2 and MB. | |
3.2. PA spectra of suspension containing MB and TiO2
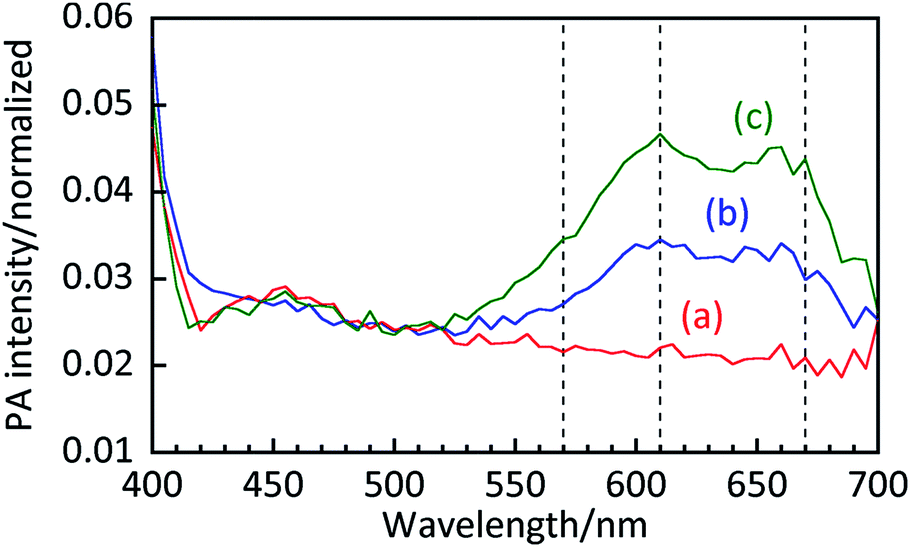
Fig. 4 shows the PA intensity as a function of probe wavelength in samples containing 20 g L−1 TiO2 with 0, 50, and 100 μmol L−1 MB. High PA intensities were observed below 410 nm in all samples, including the suspension without MB, which is likely due to bandgap absorption of TiO2. The peaks in PA intensity around 550–700 nm are attributed to MB because of a lack of peaks in that spectral region in the suspension containing no MB. Compared to an aqueous MB solution, the ratio of the absorption at 610 nm to that at 670 nm is clearly higher for the suspension. Since absorption at 610 nm is attributed to side-by-side aggregated MB dimers,29 this result indicates that MB exists both in the form of monomers and dimers on the TiO2 surface.
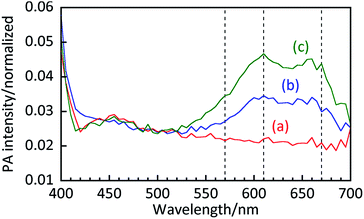 |
| | Fig. 4 PA spectra of aqueous suspensions containing 20 g L−1 of TiO2 and (a) 0, (b) 50, and (c) 100 μmol L−1 of MB. | |
Fig. 5 shows PA spectra of TiO2 powder with and without MB. For the powder sample, higher PA intensity is observed at 500–700 nm as well as below 410 nm. These are attributed to MB adsorbed on the TiO2 surface and bandgap absorption of TiO2, respectively. For the powder sample, the peak at 570 nm, attributed to photoabsorption of MB trimers,29 was observed in addition to the peaks at 610 nm and 670 nm though it was hardly observed for the suspension samples in Fig. 4. Thus, photoabsorption of dry powder is different to that in the suspension due to strong aggregation of MB on the TiO2 surface.
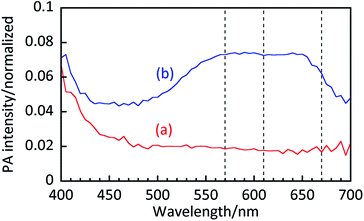 |
| | Fig. 5 PA spectra of TiO2 powders (a) without and (b) with MB. | |
3.3. Time course of MB decolorization
Fig. 6a shows time-course plots of MB concentration in the TiO2 suspension under an O2 purge. The MB concentration was calculated from the standard curve shown in Fig. 3. Before UV irradiation, the MB concentration in the suspension was constant, indicating that no MB self-degradation occurs under the probe light. This is because the probe intensity is relatively low (ca. 4.0 mW) compared with that for other reported laser-induced PAS studies (100–200 mW).24,25 The MB concentration decreased with increasing UV irradiation time, and k was calculated to be 0.075 s−1. No recovery of color was observed in the dark after complete bleaching. This indicates that an active species, such as positive hole, electron, hydroxyl radicals and/or active oxygen species, oxidizes MB into colorless products. In the presence of ethanol (Fig. 6b), the MB concentration showed little decrease because positive holes and hydroxyl radicals are quenched by ethanol. Thus, the decrease in the MB concentration observed in Fig. 6a can possibly be attributed to oxidation of MB by positive holes or hydroxyl radicals.
 |
| | Fig. 6 Time-course plots of MB concentration in suspensions initially containing 20 g L−1 TiO2 and 50 μmol L−1 MB (pH 5.6) (a) without and (b) with vol% ethanol. The cuvette headspace was thoroughly purged with O2 gas during the measurements. | |
Fig. 7a shows the time-course plots of MB concentration in the TiO2 suspensions with ethanol under a N2 purge. In the absence of O2, self-degradation of MB was again not observed before UV irradiation. Under UV irradiation, the MB concentration immediately decreased with k of 1.5 s−1 despite the presence of ethanol. It is well known that leuco-methylene blue (LMB), which is a colorless product with an absorption peak at 256 nm, is generated in the absence of oxygen via reduction through the following mechanism.9,30
| MB + H+ + 2e− = LMB, 0.532 V vs. NHE |
 |
| | Fig. 7 Time-course plots of MB concentration in the suspension containing 20 g L−1 of TiO2 and 50 μmol L−1 of initial MB (pH 5.6) (a) with 10 vol% ethanol and (b) without. The headspace of the cuvette was thoroughly purged with N2 gas for 60 min from the start of measurement, after which it was purged with O2 gas. | |
Because TiO2 has a conduction band with a more negative potential than that for reduction of MB into LMB, LMB can be generated by photocatalytic reduction over TiO2. It is reasonable that the MB bleaching rate was faster than that observed in the MB decolorization process under an O2 purge (shown above in Fig. 6a) because MB reduction is favorable due to the less negative potential than redox potential of O2 reduction (O2 + e− = O2−(aq), −0.284 V vs. NHE, O2 + H+ + e− = HO2(aq), −0.046 V vs. NHE). Therefore, the decrease in MB concentration is not attributed to oxidative MB degradation but to the reductive generation of LMB, which occurs through the quenching of positive holes and hydroxyl radicals by ethanol. A complete recovery of MB was observed in dark conditions under an O2 purge. This suggests that LMB was oxidized by ambient O2 to regenerate MB.9
| 2LMB + 2H+ + O2 → 2MB + H2O |
This recovery process explains why no decolorization due to reduction of MB is observed in Fig. 6b.
Fig. 7b shows the time-course plots of the MB concentration in the TiO2 suspension under a N2 purge. Under UV irradiation, k was calculated to be 0.44 s−1 the MB concentration in the suspension decreases faster than for normal MB degradation processes (Fig. 6a) and slower than for the suspension with ethanol (Fig. 7a). Complete bleaching was observed under UV irradiation, while an incomplete recovery of MB was observed in the dark under O2 purge. These results indicate that, under UV irradiation, some MB was reduced to LMB and some was oxidized to form colorless products. Thus, oxidation of MB proceeds not by an active oxygen species but by positive holes or hydroxyl radicals, and ambient O2 is also required to consume electrons as a counterpart to MB oxidation. These observations agree with those of previously reported studies.7,9
3.4. Initial pH dependence of MB decolorization
Fig. 8 shows the concentration of decolored MB after 30 min of UV irradiation as a function of initial pH of the suspension. Because not all time-course plots obtained at various initial pH in suspensions was fitted to a pseudo-first-order model, concentration of decolored MB after 30 min of UV irradiation was shown. Previous studies suggest that the rate of MB degradation is higher under basic conditions because cationic MB preferentially adsorb on anionic TiO2 surface.13–15 In the present study, we obtained similar results, and a decrease in decolored MB was observed with decreasing pH (for pH values lower than 5.6). This result indicates that the adsorption of MB determined the MB decolorization rate. On the other hand, decolored MB was slightly decreased with increasing pH higher than 5.6. One plausible reason for this is that strongly adsorbed MB covers the TiO2 surface such that O2 reduction, a counterpart of MB oxidation, is retarded. Moreover, a decrease in MB degradation rate with an increase in initial pH may indicate that hydroxyl radicals is not active species for MB degradation. Differences between our results for the initial pH dependence of decolored MB and the results of previous studies are possibly due to a systematic underestimation of the “true” MB concentration in the suspension by the conventional method mentioned in the introduction.
 |
| | Fig. 8 Concentration of decolored MB after 30 min of UV irradiation as a function of initial pH in suspensions initially containing 20 g L−1 TiO2 and 50 μmol L−1 MB. The headspace of the cuvette was thoroughly purged with O2 gas during the measurements. | |
4 Conclusions
The decolorization of MB, which is a conventional method for photocatalytic evaluation, was investigated using PAS in a corrosion-resistant PA cell with a microphone detector. PAS measurements enabled the detection of both “free” MB in solution and MB adsorbed onto TiO2 surfaces. This method improved upon past MB decolorization measurements because it allowed the photocatalytic activity to be measured through MB concentration (μmol L−1) instead of the final-to-initial concentration ratio, “C/C0”. Moreover, PAS enabled in situ time-course measurements of photocatalytic MB decolorization were performed at 20 s intervals. The results in this paper suggest that PAS is a powerful tool for the analysis of photocatalytic dye decolorization as well as in situ observations of photocatalytic suspension reactions.
References
- A. Fujishima, T. N. Rao and D. A. Tryk, J. Photochem. Photobiol., C, 2000, 1, 1–21 CrossRef CAS.
- M. R. Hoffmann, S. T. Martin, W. Choi and D. W. Bahnemann, Chem. Rev., 1995, 95, 69–96 CrossRef CAS.
- N. Zhang, M. Yang, S. Liu, Y. Sun and Y. Xu, Chem. Rev., 2015, 115, 10307–10377 CrossRef CAS PubMed.
- W. Ong, L. Tan, S. Chai, S. Yong and A. R. Mohamed, ChemSusChem, 2014, 7, 690–719 CrossRef CAS PubMed.
- H. Zhang, G. Liu, L. Shi, H. Liu, T. Wang and J. Ye, Nano Energy, 2016, 22, 149–168 CrossRef CAS.
- X. Yan, T. Ohno, K. Nishijima, R. Abe and B. Ohtani, Chem. Phys. Lett., 2006, 429, 606–610 CrossRef CAS.
- A. Mills and J. Wang, J. Photochem. Photobiol., A, 1999, 127, 123–134 CrossRef CAS.
- A. Mills, C. Hill and P. K. J. Robertsonc, J. Photochem. Photobiol., A, 2012, 237, 7–23 CrossRef CAS.
- A. Mills, Appl. Catal., B, 2012, 128, 144–149 CrossRef CAS.
- M. Kosmulski, Langmuir, 1997, 13, 6315–6320 CrossRef CAS.
- K. Bourikas, C. Kordulis and A. Lycourghiotis, Environ. Sci. Technol., 2005, 39, 4100–4108 CrossRef CAS PubMed.
- M. Kosmulski, J. Colloid Interface Sci., 2006, 298, 730–741 CrossRef CAS PubMed.
- C. Guillard, H. Lachheb, A. Houas, M. Ksibi, E. Elaloui and J. Herrmann, J. Photochem. Photobiol., A, 2003, 158, 27–36 CrossRef CAS.
- R. J. Tayade, T. S. Natarajan and H. C. Bajaj, Ind. Eng. Chem. Res., 2009, 48, 10262–10267 CrossRef CAS.
- A. Houas, H. Lachheb, M. Ksibi, E. Elaloui, C. Guillard and J. Herrmann, Appl. Catal., B, 2001, 31, 145–157 CrossRef CAS.
- A. Rosencwaig and A. Gersho, J. Appl. Phys., 1976, 47, 64–69 CrossRef.
- A. C. Tam, Rev. Mod. Phys., 1986, 58, 381–431 CrossRef CAS.
- T. Iwasaki, S. Oda, H. Kamada and K. Honda, J. Phys. Chem., 1980, 84, 1060–1061 CrossRef CAS.
- T. Iwasaki, S. Oda, T. Sawada and K. Honda, J. Phys. Chem., 1980, 84, 2800–2802 CrossRef CAS.
- T. Sawada, S. Oda, T. Iwasaki, K. Honda and H. Kamada, Proc. Jpn. Acad., Ser. B, 1980, 56, 649–653 CrossRef CAS.
- N. Murakami, O. O. P. Mahaney, T. Torimoto and B. Ohtani, Chem. Phys. Lett., 2006, 426, 204–208 CrossRef CAS.
- N. Murakami, O. O. P. Mahaney, R. Abe, T. Torimoto and B. Ohtani, J. Phys. Chem. C, 2007, 111, 11927–11935 CAS.
- S. Morishita, A. Fujishima and K. Honda, J. Electroanal. Chem. Interfacial Electrochem., 1983, 143, 433–438 CrossRef CAS.
- Y. Nosaka, R. Igarashi and H. Miyama, Anal. Chem., 1985, 57, 92–94 CrossRef CAS.
- S. Oda, T. Sawada, T. Moriauchi and H. Karnada, Anal. Chem., 1980, 52, 650–652 CrossRef CAS.
- C. K. N. Patel and A. C. O. Tam, Appl. Phys. Lett., 1979, 34, 467–469 CrossRef CAS.
- A. C. Tam and C. N. K. Patel, Opt. Lett., 1980, 5, 27–29 CrossRef CAS PubMed.
- S. Oda and T. Sawada, Anal. Chem., 1981, 53, 471–474 CrossRef CAS.
- B. Liu, L. Wen, K. Nakata, X. Zhao, S. Liu, T. Ochiai, T. Murakami and A. Fujishima, Chem.–Eur. J., 2012, 18, 12705–12711 CrossRef CAS PubMed.
- O. Impert, A. Katafias, P. Kita, A. Mills, A. Pietkiewicz-Graczyk and G. Wrzeszcz, Dalton Trans., 2003, 348–353 RSC.
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.