
Aminolysis of triglycerides using nanocrystalline nickel doped CaO as an efficient solid catalyst†
Dinesh Kumar ab,
KM Abidaa and
Amjad Ali*a
ab,
KM Abidaa and
Amjad Ali*a
aSchool of Chemistry and Biochemistry, Thapar University, Patiala-147004, India. E-mail: amjadali@thapar.edu; amjad_2kin@yahoo.com; Fax: +91-175-2393005; Tel: +91-175-2393832
bKU-KIST Graduate School of Science & Technology, Korea University, Seoul, South Korea-136-701
First published on 30th June 2016
Abstract
The present manuscript reports the preparation of Ni doped CaO in nanocrystalline form, using a wet chemical method, and the investigation of their catalytic activity towards aminolysis of a variety of triglycerides with diethanolamine. The powder XRD study suggested the formation of a solid solution of NiO in CaO as long as the nickel content in the catalyst remained ≤5 wt%. A transmission electron microscopy study supported the formation of Ni/CaO particles in the form of nanorods at a 650 °C calcination temperature. Catalyst activity was found to be a function of basic strength which in turn depends upon the catalyst calcination temperature. The catalyst prepared by 0.5 wt% Ni doping in CaO at 650 °C was found to complete the aminolysis (>99% amide yield) of waste cottonseed oil within 0.5 and 3.0 h at 110 °C and room temperature (35 °C), respectively. The catalyst was easily recovered from the reaction mixture by simple filtration and reused during seven consecutive reaction cycles without a significant loss in activity. The (pseudo) first order rate constant and activation energy for the reaction was observed as 0.15 min−1 and 52.7 kJ mol−1, respectively.
1. Introduction
Renewable raw materials viz., vegetable oils and fat are going to play a significant role in the development of sustainable green chemistry.1 Derivatives of vegetable oil have abundant benefits as they are non-toxic, renewable, biodegradable and eco-friendly. These derivatives have a variety of commercial applications such as fatty acid methyl ester, commonly known as biodiesel, which is a nontoxic, biodegradable, eco-friendly and renewable substitute for conventional fossil fuel2 and fatty acid amides have a wide range of application as surfactants, lubricants, cosmetics, shampoos, foam control agents, fungicides, corrosion inhibitors, water repellents, detergents and anti-blocking agents in the plastics processing industry.3–5 They are less reactive and hence, employed as anti-slip and anti-block additives in polyethylene films and also as flow improvers.6 Fatty acid amides also improve the ignition quality of diesel fuel and for this reason they may be employed as a cetane number improver for diesel fuel.7,8 Amides are conventionally prepared by reacting amines with carboxylic acid or its more reactive derivatives9,10 in the presence of metal based catalysts11–13 or bio-catalysis or under microwave irradiation.14–19 At the industrial scale, fatty amides of triglycerides are synthesized in a two step reaction process20 involving (i) the conversion of triglyceride into a fatty acid ester followed by (ii) aminolysis of esters to obtain the corresponding amide derivative as shown in Fig. S1 (ESI†).Direct or one step aminolysis of vegetable oils is not common, though it can be performed in the presence of biocatalysts21–23 or microwave irradiation in combination with a homogeneous catalyst.24 A one step method may be advantageous over a conventional method as it would generate fewer by-products and require fewer purification steps. For the aminolysis of esters and carboxylic acids, in the literature, indium triiodide,25 group (IV) metal alkoxide complexes,26 CdO and SnCl2 in ionic medium were employed. The use of a homogeneous catalyst for the synthesis of fatty acid amides generates a contaminated product and hence, a tedious purification step is inevitable which generates a large amount of effluents.27 Due to increasing environmental and economic concerns, invention and application of green and cost effective processes have become a main concern for the researchers working in the area of fatty acid amide synthesis.28 In this regard, the use of heterogeneous catalysts, due to reusability and ease of separation, for amide synthesis may be beneficial.
The literature reported bio- as well as chemical heterogeneous catalysts such as Novozym 435 (using 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 of amine:feedstock molar ratio),29 sodium methoxide,30 sodium methylate,31 sodium ethoxide,32 ZnO·La2CO5·LaOOH,20 CdO (with an ionic liquid as support)27 either show incomplete conversion and/or required long reaction time (up to 24 h) to obtain satisfactory conversion levels (>90%). To the best of our knowledge, there is no reported work available in the literature employing Ni doped CaO (Ni/CaO) as a heterogeneous catalyst for the aminolysis of triglycerides or fatty acid methyl esters.
:1 of amine:feedstock molar ratio),29 sodium methoxide,30 sodium methylate,31 sodium ethoxide,32 ZnO·La2CO5·LaOOH,20 CdO (with an ionic liquid as support)27 either show incomplete conversion and/or required long reaction time (up to 24 h) to obtain satisfactory conversion levels (>90%). To the best of our knowledge, there is no reported work available in the literature employing Ni doped CaO (Ni/CaO) as a heterogeneous catalyst for the aminolysis of triglycerides or fatty acid methyl esters.
The present manuscript demonstrates the preparation of Ni/CaO in nanosized form by a simple chemical method and its characterization by SEM, TEM, powder XRD, and Hammett indicator studies. The prepared Ni/CaO was employed as a heterogeneous catalyst for the aminolysis of triglycerides with diethanolamine to prepare the corresponding amide derivatives. The same catalyst was also successfully employed for the transesterification of triglycerides to prepare fatty acid methyl esters (FAME) and for further aminolysis of FAME to produce fatty acid amides.
2. Experimental section
2.1 Materials and methods
Waste cotton seed oil was collected from the restaurants situated in Patiala. Nickel nitrate, CaO, methanol (99.8%) and diethanolamine (99%) were purchased from Merck, India. Methyl laurate (99%) was procured from Sigma-Aldrich, USA.Scanning electron microscopy (SEM) images were collected on a JEOL JSM 6510LV and transmission electron microscopy (TEM) images on HITACHI 7500 instruments. Scanning Microscopy-Energy Dispersive X-ray analysis (SEM-EDX) has been performed for the qualitative analysis of the catalysts. Fourier transform-nuclear magnetic resonance (FT-NMR) of organic molecules were recorded on a Bruker Avance-II (400 MHz) spectrophotometer using CDCl3 as the solvent and tetramethylsilane (TMS) as an internal reference. The presence of amide functional groups have been supported with the help of FTIR spectra recorded on a Thermo Scientific Nicolet iS10 spectrometer in ATR mode. The free fatty acid (FFA), saponification, iodine value and moisture content of the virgin soybean oil (SO), virgin cotton seed oil (CO), mutton fat (MF), waste cottonseed seed oil (WO), karanja oil (KO) and jatropha oil (JO), were determined by following the literature reported methods33 and corresponding observed values are provided in Table 1.
| Feedstock | Free fatty acid value (wt%) | Moisture content (wt%) | Saponification value (mg KOH per g) | Iodine value |
|---|---|---|---|---|
| Waste cotton seed oil | 1.8 | 0.3 | 190.4 | 101.7 |
| Karanja oil | 4.3 | 0.3 | 185.0 | 96.8 |
| Jatropha oil | 8.4 | 0.4 | 195.5 | 101.5 |
| Virgin cotton seed oil | 0.1 | 0.3 | 188.7 | 102.6 |
| Virgin soybean oil | 0.2 | 0.3 | 190.0 | 125.4 |
| Mutton fat | 0.9 | 0.3 | 192.4 | 46.1 |
The basic strength of the catalysts (pKBH+) was measured by the Hammett indicator benzene carboxylic acid titration method,34 employing neutral red (pKBH+ = 6.8), bromothymol blue (pKBH+ = 7.2), phenolphthalein (pKBH+ = 9.3), Nile blue (pKBH+ = 10.1), tropaeolin-O (pKBH+ = 11.1), 2,4-dinitroaniline (pKBH+ = 15.0), and 4-nitroaniline (pKBH+ = 18.4) as indicators.
2.2 Preparation of the catalyst
A series of Ni/CaO catalysts were prepared by a wet chemical method in which, CaO (10 g) was suspended in 40 mL deionized water and to this desired amount of Ni(NO3)2·6H2O aqueous solution was added to obtain 0.5–6 wt% nickel content in CaO. The resulted slurry was stirred for 3 h, dried and then calcined in the temperature range of 150–950 °C for 12 h. The prepared catalysts were designated as x-Ni/CaO-T, where x and T is the nickel content (wt%) in CaO, and calcination temperature, respectively.2.3 Catalyst activity
The prepared Ni/CaO catalysts were employed for the aminolysis reaction of vegetable oil as well as FAME in order to obtain the amide derivative and transesterification of vegetable oils to obtain FAME as demonstrated in Scheme 1. Thus the Ni/CaO catalyst can be employed for the FAA synthesis either in a single step via direct aminolysis of triglycerides or in a two step process involving (i) transesterification of triglycerides to obtain FAME followed by (ii) the aminolysis of FAME to yield FAA. During the one step process, glycerol is obtained as a by-product which is cumbersome to remove from the FAA owing to the high boiling point of both compounds (>300 °C). To avoid this problem, a two step process may be preferred for FAA synthesis as glycerol, produced during the first step, is immiscible with FAME and hence, could be easily recovered from the reaction mixture. The detailed experimental procedure for all reactions is as follows: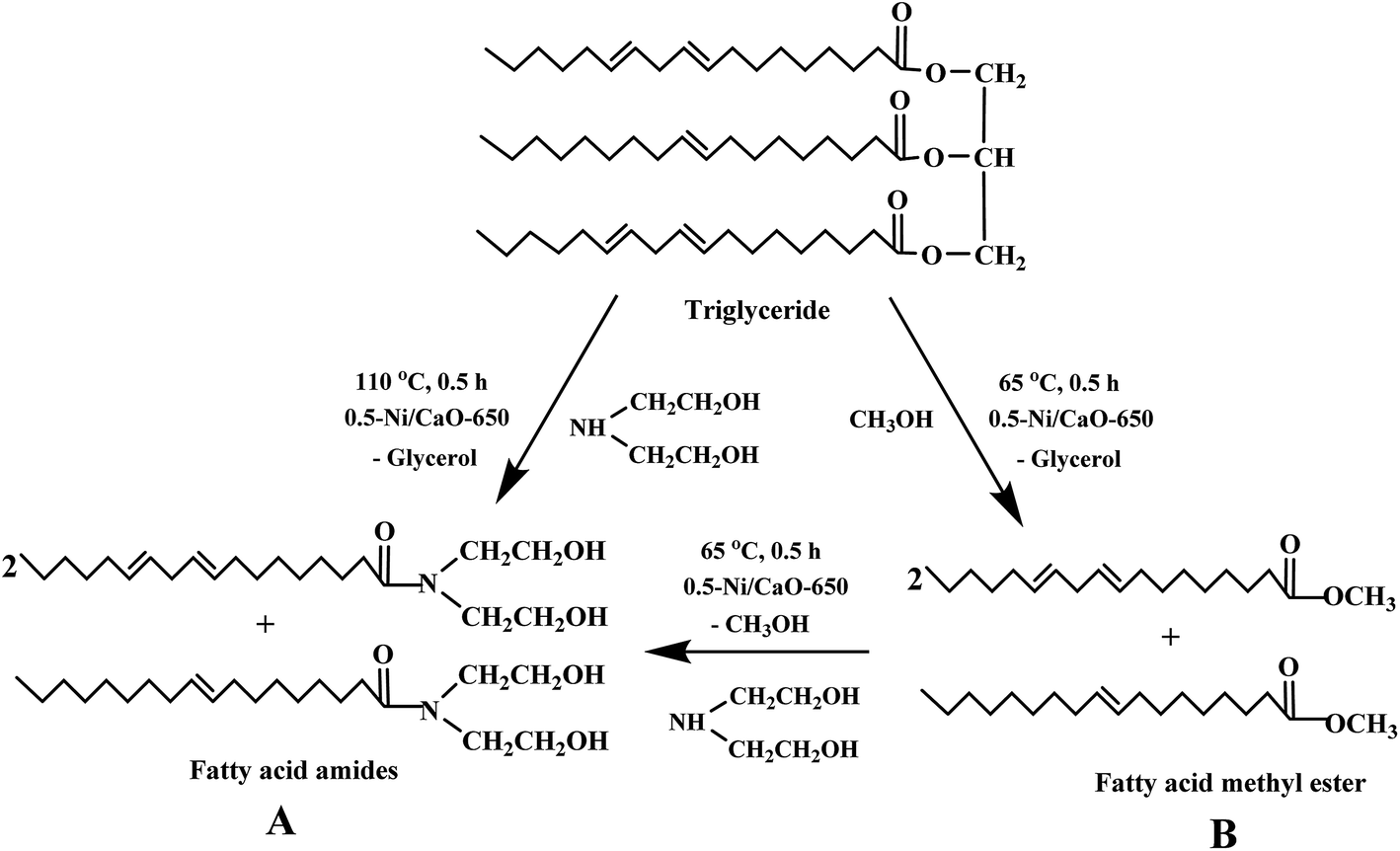 | ||
| Scheme 1 Ni/CaO catalyzed (A) aminolysis and (B) transesterification reactions. | ||
The reaction progress was monitored, after regular time intervals, by withdrawing a small amount of the reaction mixture with the help of a glass capillary and subjecting it to the FTIR analysis.
:1 molar concentration of methanol to oil, and 0.5 g of the 0.5-Ni/CaO-650 catalyst were heated at 65 °C for 30 min. Upon completion of the reaction, the catalyst was recovered by centrifugation and the liquid phase was kept overnight in a separating funnel to separate the upper FAME layer from the lower glycerol layer. Excess alcohol from FAME was recovered with the help of a rotary evaporator and finally FAME was quantified by a proton NMR technique. The prepared FAME was employed for the aminolysis as shown in reaction Scheme 1.The amide derivative produced during the aminolysis reaction was characterized by FTIR and proton NMR and quantified by FTIR spectroscopy.8 FAME prepared by transesterification of vegetable oils and mutton fat has been characterized and quantified by proton NMR spectroscopy,35 respectively.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–) in both molecules.
CH–) in both molecules.
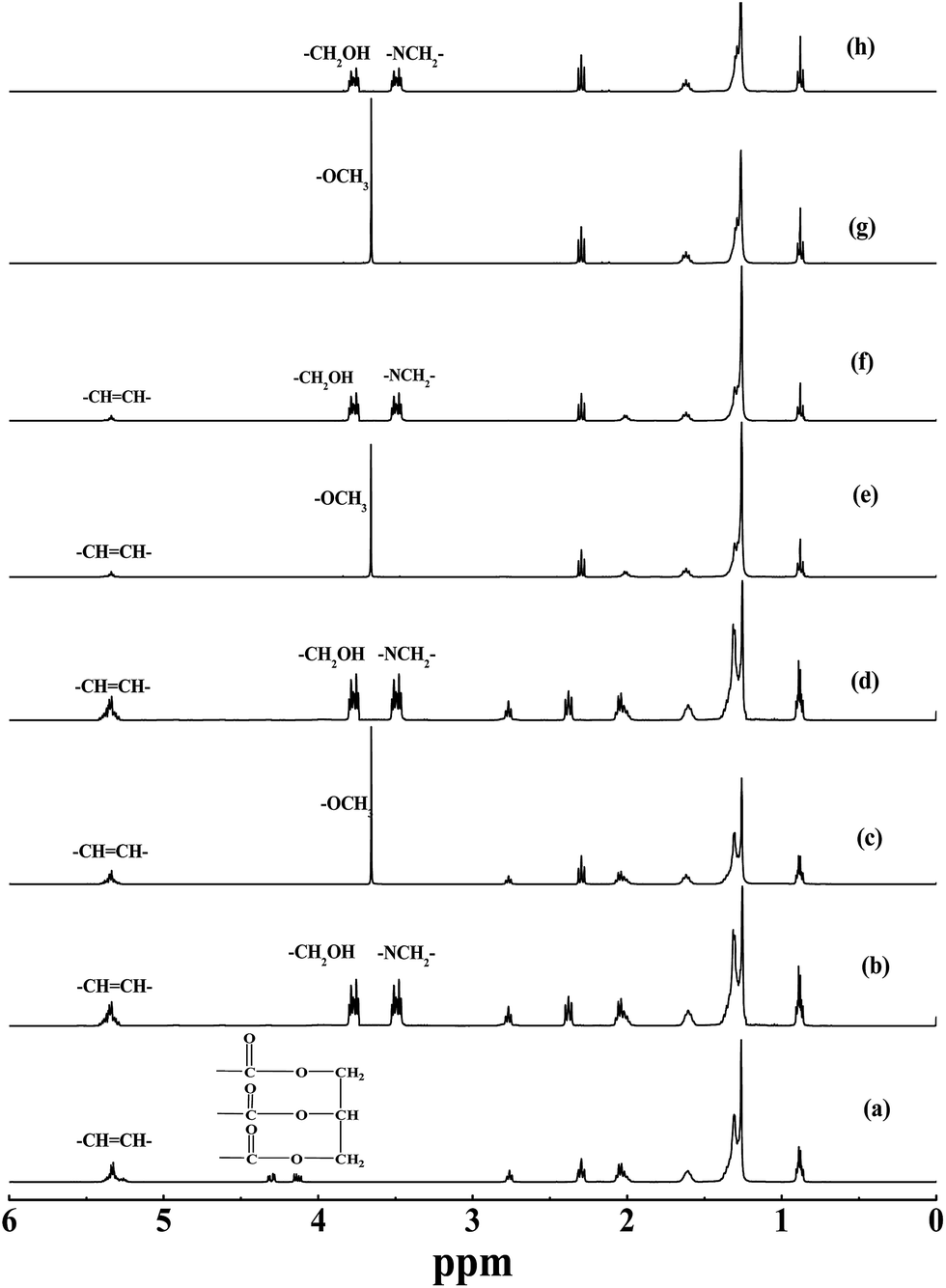 | ||
| Fig. 1 Comparison of 1H-NMR spectra of (a) cotton seed oil, (b) the fatty acid amide of cotton seed oil, (c) cotton seed oil FAME, (d) the fatty acid amide of cotton seed oil derived FAME, (e) mutton fat FAME, (f) the fatty acid amide of mutton fat derived FAME, (g) methyl laurate, and (h) the amide derivative of methyl laurate. | ||
NMR and FTIR data for FAME and FAA prepared in the presence of the Ni/CaO catalyst is given in the ESI.†
3. Results and discussion
3.1 Catalyst characterization
The activity of CaO based catalysts was found to be a function of its basic strength. As shown in Table 2, pure CaO demonstrated low basic strength as well as poor aminolysis activity. The basic strength as well as CaO activity was tuned by gradually increasing the Ni2+ content (0.5–6 wt%) as well as the calcination temperature (150 to 950 °C). Initially the effect of the Ni2+ content on the catalyst structure and activity was studied at a fixed calcination temperature of 650 °C. As long as the nickel content in the catalyst remained ≤5 wt%, no peaks corresponding to NiO was observed in the powder XRD patterns of Ni/CaO (Fig. 2). This could be attributed to the high degree of dispersion of Ni2+ in CaO. However, on increasing the nickel concentration up to 6 wt%, low intensity peaks at 2θ values of 43.2° and 62.7° were observed to indicate the presence of NiO in the cubic phase (JCPDS 780643) as shown in Fig. 2. Hence, a maximum of 5 wt% Ni2+ could be doped without affecting the regular CaO structure. As could be seen from Table 2, no significant increase in basic strength was observed on increasing the nickel concentration in the catalyst beyond 0.5 wt%. Hence, 0.5 wt% Ni2+ was considered as the optimum amount for the improved catalytic activity of Ni/CaO.| Catalyst type | Basic strength (pKBH+) | Total basicitya (mmol g−1 of catalyst) | BET surface area (m2 g−1) | TOFb (h−1) |
|---|---|---|---|---|
| a Measured by Hammett indicator titration method using indicators in the pKBH+ range of 6.8 to 11.1.b Rate of reaction/mmol of catalyst active site, NM = not measured. | ||||
| Commercial-CaO | 9.8 < pKBH+ < 10.1 | 7.5 | 3.9 | 1.3 |
| 0.5-Ni/CaO-150 | 11.1 < pKBH+ < 15 | 8.6 | 3.8 | 4.6 |
| 0.5-Ni/CaO-250 | 11.1 < pKBH+ < 15 | 8.7 | 3.8 | 5.7 |
| 0.5-Ni/CaO-350 | 11.1 < pKBH+ < 15 | 8.9 | 4.8 | 7.4 |
| 0.5-Ni/CaO-450 | 11.1 < pKBH+ < 15 | 9.4 | 12.9 | 10.5 |
| 0.5-Ni/CaO-650 | 15 < pKBH+ < 18.4 | 9.9 | 14.7 | 20.0 |
| 0.5-Ni/CaO-850 | 15 < pKBH+ < 18.4 | 10.1 | 11.7 | 6.5 |
| 0.5-Ni/CaO-950 | 11.1 < pKBH+ < 15 | 12.3 | 3.9 | 4.0 |
| 1-Ni/CaO-650 | 15 < pKBH+ < 18.4 | 12.4 | NM | 15.9 |
| 2-Ni/CaO-650 | 15 < pKBH+ < 18.4 | 12.2 | NM | 16.2 |
| 3-Ni/CaO-650 | 15 < pKBH+ < 18.4 | 12.4 | NM | 15.9 |
| 4-Ni/CaO-650 | 15 < pKBH+ < 18.4 | 12.2 | NM | 16.2 |
| 5-Ni/CaO-650 | 15 < pKBH+ < 18.4 | 12.2 | NM | 16.2 |
| 6-Ni/CaO-650 | 15 < pKBH+ < 18.4 | 12.3 | NM | 16.1 |
 | ||
| Fig. 2 Comparison of XRD diffraction patterns of CaO doped with 0.5–6 wt% nickel (* = calcium oxide and ● = nickel oxide). | ||
In order to optimize the calcination temperature, 0.5 wt% Ni2+ doped CaO was calcined in the temperature range of 150 to 950 °C. As indicated in the XRD patterns (Fig. 3), up to 350 °C calcination temperature Ca(OH)2 was found as a major phase. The formation of Ca(OH)2 is attributed to the reaction of CaO with water which is used as the reaction medium during the catalyst preparation. A minor phase of CaCO3 was also observed due to the reaction of atmospheric CO2 with CaO, as evident from the XRD patterns of the samples calcined up to 550 °C. At 450 °C, diffraction patterns corresponding to the CaO phase were observed due to the partial thermal decomposition of Ca(OH)2. Nevertheless, above 550 °C calcination temperature due to the complete decomposition of Ca(OH)2 as well as CaCO3, a single CaO phase was observed in the XRD patterns of 0.5-Ni/CaO-650 and subsequent samples (Fig. 3). The calcination process transformed the Bronsted basic sites (–OH) into Lewis basic sites (–O–), which were found to be more active during the transesterification as well as aminolysis reaction. Since basic strength as well as activity were found to be maximal at 650 °C, a detailed study was performed with the catalyst prepared at 650 °C.
 | ||
| Fig. 3 Comparison of XRD spectra of commercially available CaO with 0.5-Ni/CaO-150, 0.5-Ni/CaO-350, 0.5-Ni/CaO-650 and 0.5-Ni/CaO-950 (* = calcium oxide, ◆ = calcium hydroxide, and □ = calcium carbonate). | ||
The BET surface area of pure calcium oxide and Ni/CaO is compared in Table 2. An increase in surface area from 4.8 to 14.7 m2 g−1 at 350 to 650 °C calcination temperature could be attributed to the thermal decomposition of Ca(OH)2 as well as simultaneous removal of water molecules from the catalyst. Water molecule exclusion left voids in catalyst particles, increasing the surface area of the catalyst. A further increase in calcination temperature (750 to 950 °C) was found to reduce the surface area due to sintering of smaller particles into relatively larger particles.
The N2 adsorption–desorption isotherms of 0.5-Ni/CaO-350, 0.5-Ni/CaO-650 and 0.5-Ni/CaO-950 are shown in Fig. S3A (ESI†). For all three samples, the sharp steps of capillary condensation of N2 are observed at P/P0 = 0.8–0.9, to indicate a typical pattern IV of IUPAC classification and H1-type hysteresis loops. Furthermore, in the case of 0.5-Ni/CaO-350 and 0.5-Ni/CaO-950, the hysteresis loop becomes narrower and for 0.5-Ni/CaO-650 the loop becomes broader. This observation further supports the formation of smaller sized pores in the samples calcined at a 350 and 950 °C calcination temperature and relatively larger sized pores at a 650 °C calcination temperature36,37
The narrow BJH pore-size distribution curves of 0.5-Ni/CaO-350 and 0.5-Ni/CaO-950 confirm that the pore size distribution is uniform for both the materials. However, broad pore-size distribution curves for 0.5-Ni/CaO-650 (Fig. S3B, ESI†) signify the less uniform pore-size distribution for this sample.
The solid state UV-visible diffuse reflectance spectra of pure CaO and 0.5-Ni/CaO-650 is compared in Fig. S4 (ESI†). A shoulder at 210 nm was observed in CaO due to the O2− → Ca2+ charge transfer, while the presence of a strong band at 254 nm in the case of Ni/CaO could be attributed to the O2− → Ni2+ charge transitions.38–40 However, no d → d band was observed in the visible region mainly due to the small amount of Ni2+ in CaO.
The particle size and surface morphology of 0.5-Ni/CaO, prepared at 350, 650 and 950 °C calcination temperature, were observed by scanning and transmission electron microscopy (Fig. 4). At 350 °C, the SEM as well as TEM study supported the formation of agglomerates of hexagonal, cubic and oval shaped particles (Fig. 4a and b). At 650 °C, no significant difference was observed by SEM (Fig. 4c), however, TEM images indicate the formation of nanorods of Ni/CaO with an average length of ∼100 nm and a thickness of ∼10 nm (Fig. 4d). Thus TEM analysis also supports the high surface area observed at 650 °C calcination temperature. At 950 °C, the SEM as well as TEM study support the formation of Ni/CaO agglomerates mainly due to the sintering of the particles (Fig. 4e and f). SEM-EDX analysis supported the presence of 0.6% nickel in 0.5-Ni/CaO-650 particles.
 | ||
| Fig. 4 Comparative FESEM images of (a) 0.5-Ni/CaO-350, (c) 0.5-Ni/CaO-650, and (e) 0.5-Ni/CaO-950 and TEM images of (b) 0.5-Ni/CaO-350, (d) 0.5-Ni/CaO-650, and (f) 0.5-Ni/CaO-950. | ||
3.2 Catalytic activity
To examine the aminolysis activity of the Ni/CaO catalyst, waste cotton seed oil along with diethanolamine was employed as the substrate. In order to establish the optimum condition to achieve >99% conversion levels, the following parameters were varied during the course of the study: (i) Ni2+ content in CaO (ii) calcination temperature, (iii) catalyst concentration, (iv) reaction temperature and (v) diethanolamine/oil molar ratio.:1 diethanolamine/oil molar ratio) at 110 °C in the presence of 5 wt% prepared catalysts. The reaction time required for complete aminolysis was found to be decreased from 1 h to 0.5 h as the amount of nickel ion in CaO was increased from 0.25 to 0.5 wt%. However, a further increase in Ni2+ concentration does not reduce the reaction time to a significant extent as shown in Fig. 5A. Hence, the 0.5-Ni/CaO-650 catalyst was selected for optimizing other parameters to achieve the minimum time for the complete aminolysis reaction.
 | ||
| Fig. 5 Effect of (A) Ni2+ content and (B) calcination temperature on reaction duration to achieve >99% amide yield (reaction conditions: diethanolamine:waste cotton seed oil = 5:1 (m/m), catalyst amount = 5 wt% of feedstock and temperature = 110 °C). | ||
In order to determine the optimum calcination temperature for the prominent catalytic activity, a series of catalysts were prepared by doping 0.5 wt% Ni2+ in CaO and calcining the same in the temperature range of 150–950 °C. A 5 wt% amount of the catalysts with respect to oil has been used for the aminolysis reaction of waste cotton seed oil with diethanolamine (1:5 molar ratio) at 110 °C. The reaction time required for the complete aminolysis was found to decrease from 2.5 h to 0.5 h as the calcination temperature increases from 250 to 650 °C. As discussed in the XRD study, calcination of Ni/CaO causes the thermal decomposition of Bronsted basic sites (Ca(OH)2) into Lewis basic sites (CaO). In the present study, aminolysis activity by Lewis basic sites was found to be better than the activity of Bronsted sites of Ca(OH)2 towards the same reaction as reported in our earlier work.41,42 A further increase in calcination temperature reduces the catalyst activity as more time was required by the catalyst for the completion of the reaction as shown in Fig. 5B. This could be attributed to the decrease in surface area due to the sintering of catalyst particles which is accompanied by the decrease in active sites at the catalyst surface. Hence, a catalyst having 0.5 wt% Ni2+ content CaO and prepared at a 650 °C calcination temperature (0.5-Ni/CaO-650) was selected for optimizing other parameters for the aminolysis of waste cotton seed oil.
:1 molar ratio) at 110 °C were performed in the presence of nanocrystalline 0.5-Ni/CaO-650 by varying its concentration in the range of 2–7 wt% (catalyst/oil). The time required for the complete conversion decreases from 2.5 h to 0.5 h as the catalyst concentration was increased from 2 to 5 wt%. A further increase in catalyst concentration does not reduce the reaction time significantly as shown in Fig. 6A. The aminolysis reactions were further studied with 5 wt% (catalyst/oil) catalyst concentration to optimize the other parameters.
 | ||
| Fig. 6 Effect of various reaction parameters to achieve >99% amide yield (A) catalyst concentration, (B) reaction temperature, (C) diethanolamine:oil molar ratio and (D) free fatty acids (variety of feedstocks) (reaction conditions; diethanolamine:feedstock = 5:1 (m/m), catalyst (0.5-Ni/CaO-650) amount = 5 wt% of feedstock, and temperature = 110 °C). | ||
A series of aminolysis reactions were conducted in the presence of 5 wt%, (catalyst/oil) nanocrystalline 0.5-Ni/CaO-650, to find the optimum temperature for the aminolysis reaction. Time required for the complete aminolysis of vegetable oils to fatty acid amides decreases from 3 h to 0.5 h as the reaction temperature was increased from room temperature (35 °C) to 110 °C. A further increase in reaction temperature does not reduce the reaction time significantly as shown in Fig. 6B and hence, all aminolysis reactions were performed at 110 °C.
To determine the optimum diethanolamine/oil molar ratio in the present study, reactions were performed with 3:1 to 7:1 diethanolamine/oil molar ratios at 110 °C using 5 wt% of the 0.5-Ni/CaO-650 catalyst. The rate of the aminolysis reaction increases as the diethanolamine/oil molar ratio was increased from 3:1 to 5:1 and the reaction was found to be complete in 30 min when a 5:1 diethanolamine/oil molar ratio was used. A further increase in the diethanolamine/oil molar ratio does not decrease the reaction time as shown in Fig. 6C.
The presence of free fatty acids was found to reduce the catalyst efficacy due to the strong interaction between the catalyst basic sites and carboxylic group of fatty acids. The presence of FFA in waste cooking oil and nonedible oils is inevitable and hence it is worth to study the FFA tolerance of the prepared solid catalyst. In order to determine the maximum FFA tolerance of the prepared catalyst, a series of aminolysis reactions were performed with a variety of naturally occurring feedstock having 0.9–8.4 wt% FFA content. As shown in Fig. 6D, the catalyst was found to be effective for the complete aminolysis of substrates having a FFA content as high as 8.4 wt%. However, the presence of FFA was found to reduce the catalytic activity as aminolysis of karanja and jatropha oil required relatively longer reaction duration.
3.3 Aminolysis of various triglycerides and FAME
As shown in Scheme 1, the prepared catalyst could be employed either (i) for the direct aminolysis of triglycerides or (ii) in a two step reaction viz., transesterification of triglyceride followed by the aminolysis of FAME. The advantage of the first method is formation of the desired product in a single step. Nevertheless, amide and glycerol are miscible and their separation is cumbersome owing to their high boiling points. In the second method, initially, triglyceride transesterification was performed in the presence of the Ni/CaO catalyst to obtain FAME and glycerol, which were separated in a separating funnel due to the substantial difference in their density. FAME thus produced were characterized by the proton NMR technique and the corresponding spectra are given in the ESI (Fig. S5†). In the second step, FAME was employed for aminolysis in the presence of the Ni/CaO catalyst to obtain the fatty acid amide along with methanol as a by-product. Methanol, being a low boiling liquid, could be removed easily from the product with the help of a rotary evaporator. Product characterization by NMR and FTIR methods supported the formation of pure fatty acid amides in the presence of the Ni/CaO catalyst, irrespective of the method employed for the reaction. Thus, the prepared catalyst demonstrates a versatile mode of action as it is able to catalyze the (i) aminolysis of triglycerides (Fig. 6D), (ii) aminolysis of FAME (Fig. 7A) and (iii) transesterification of triglycerides (Fig. 7B). | ||
| Fig. 7 Efficacy of the 0.5-Ni/CaO-650 catalyst towards, (A) aminolysis of various FAME and (B) transesterification of various vegetable oils, to achieve >99% conversion levels (reaction conditions: aminolysis (A), diethanolamine:feedstock = 5:1 (m/m), catalyst amount = 5 wt% of feedstock, and temperature = 110 °C; transesterification (B): methanol:feedstock = 9:1 (m/m), catalyst amount = 5 wt% of feedstock, and temperature = 65 °C. Acronyms: SO = soybean oil, MF = mutton fat, WO = waste cotton seed oil, CO = castor oil, KO = karanja oil, JO = jatropha oil, SFAME = SO derived FAME, MFAME = MF derived FAME, WFAME = WO derived FAME, CFAME = CO derived FAME, KFAME = KO derived FAME, and JFAME = JO derived FAME). | ||
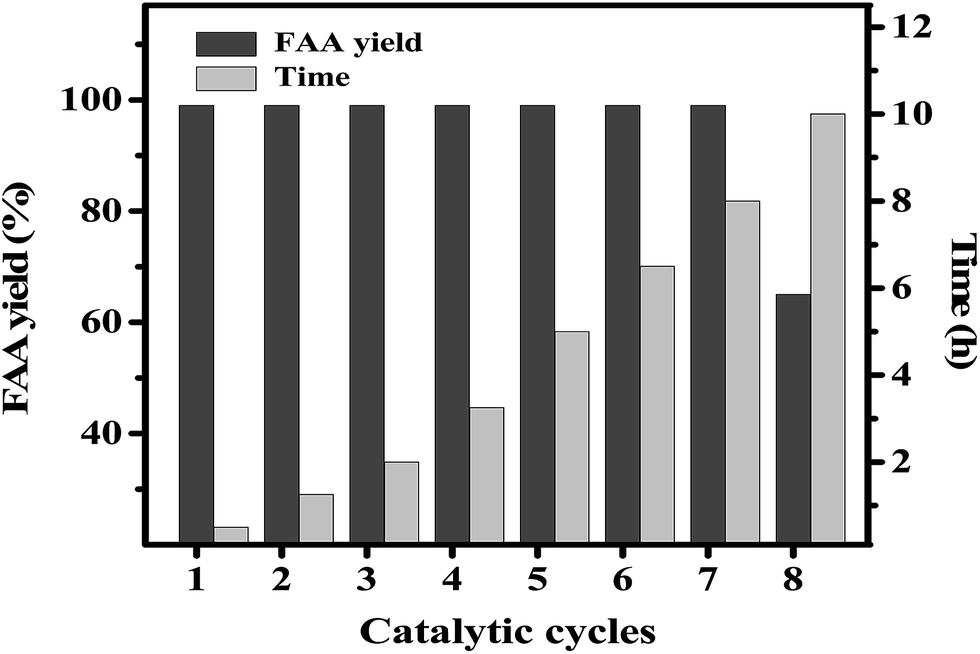 | ||
| Fig. 8 Recyclability analysis of the 0.5-Ni/CaO-650 catalyst during the aminolysis of WO (reaction conditions: diethanolamine:feedstock = 5:1 (m/m), catalyst amount = 5 wt% of feedstock, and temperature = 110 °C). | ||
The gradual loss in activity may be due to the partial dissolution of the catalyst in the reaction mixture. The dissolved catalyst may catalyze the reaction similar to a homogeneous catalyst and hence, it is vital to quantify the contribution due to the dissolved catalyst in overall catalytic activity. In order to quantify the homogeneous contribution, 0.5-Ni/CaO-650 catalyst (500 mg) has been refluxed with diethanolamine (10 g) for 0.5 h at 110 °C. After the stipulated time the catalyst was separated by centrifugation and WO oil was added to the diethanolamine (diethanolamine/oil = 5:1; m/m) and the reaction was continued at 110 °C. Product analysis after 0.5 h suggests negligible amide formation to rule out any significant contribution due to the dissolved catalyst.
In order to rationalize the cause for the loss of catalyst activity, XRD patterns of fresh and reused catalysts were compared as shown in Fig. S6 (ESI†). The XRD pattern of the reused Ni/CaO catalyst supported that structural changes have occurred during the catalyst regeneration process. The intensity of the original peaks at 2θ ∼ 37.36°, 54.36° and 32.57°, corresponding to the CaO cubic phase, was found to decrease in the reused catalyst and new peaks appeared at 25.4°, 27.4°, 36.1°, 41.3°, 48.07°, 56.7° and 62.7° to indicate the formation of a new unknown phase. Thus, upon the repeated use and regeneration process, the catalyst structure changes which ultimately cause the loss in catalytic activity.
:1 in the presence of 5 wt% catalyst (0.5-Ni/CaO-650) at 35–110 °C. The samples from the reaction mixture have been withdrawn after every 10 min, centrifuged to remove the solid catalyst, heated under vacuum to remove excess diethanolamine, and finally subjected to FTIR analysis to calculate the FAA yield. As the reaction progresses, due to the conversion of oil to amide, the intensity of the ester carbonyl band at 1743 cm−1 regularly decreases, while the intensity of the amide carbonyl peak at 1619 cm−1 increases, as shown in Fig. S7 (ESI†).The amide conversion was fitted in (pseudo) first-order rate law (i) and the linear nature of −ln(1 − Xme) vs. t (time) plots (Fig. 9A) supported that the Ni/CaO catalyzed reaction has followed (pseudo) first order kinetics.
| k = −ln{(1 − Xme)/t} | (i) |
 | ||
| Fig. 9 (A) Plot of −ln(1 − Xme) vs. time at different reaction temperatures for the 0.5-Ni/CaO-650 catalyzed aminolysis of WO; (B) the Arrhenius plot for the aminolysis of waste cotton seed oil. Reaction conditions: diethanolamine:WO = 5:1 (m/m) and catalyst amount = 5 wt%. | ||
The rate constants were obtained from the slope of these plots and found to be 0.154, 0.04, 0.028 and 0.007 min−1 at 110, 95, 65 and 35 °C reaction temperature, respectively.
The Arrhenius model43 was employed to estimate the activation energy (Ea) and pre-exponential factor (A) for the same reaction as given in eqn (ii):
|
lnk = −Ea/RT + lnA
| (ii) |
k is shown in Fig. 9B and the values of Ea and A from the graph were found to be 52.7 kJ mol−1 and 1.6 × 109 min−1, respectively. The observed activation energy was found to be >25 kJ mol−1, to indicate that the Ni/CaO catalyzed reaction is chemically controlled and not by mass transfer limitations.44
:1), (v) is reusable during seven catalytic cycles, (vi) is effective even in the presence of high FFA and (vii) is also able to catalyse the transesterification of triglycerides and aminolysis of triglycerides as well as fatty acid methyl esters.
| Catalyst | Substrate | Reaction conditions | Reference | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Catalyst amount (wt% of substrate) | Reaction temperature (°C) | Amine | Amine:substrate (m/m) |
Reaction time | FAA yield (%, m/m) | Catalyst reusability (number of reaction cycles) | |||
| a Homogeneous catalyst.b Reaction was performed in a closed vessel, MW = microwave.c The yield was decreased from 82 to 64% during reusability, p = reaction was performed at 125 mm Hg pressure, BB = benzyl bromide, NME = N-methyl ethanolamine, DEA = diethanolamine, MEA = monoethanolamine, m/m = mole/mole, NMG = N-methyl-glucamine, NA = not applicable, NR = not reported, PR = present report. | |||||||||
| Withoutb catalyst/MW | Acetic acid | NA | 100 | Dodecylamine | 30:1 |
180 s | 100 | NA | 15 |
| Withoutb catalyst/MW | Carboxylic acid | NA | 150 | n-Octylamine | 1:1 |
30 min | 3 | NA | 19 |
| Novozym 435 | Lesquerolic acid | NR | 80 | NH3 | 3:1 |
1 day | 95 | NR | 22 |
| Novozym 435 | Oleic acid | 30 | 65 | MEA | 10:1 |
3 h | 96.6 | NR | 29 |
| Novozym | Oleic acid | NR | 90 | NMG | 1:1 |
50 h | 97 | NR | 45 |
| Imidazolea/MW | Hexanoic acid | 4.5 | 100 | Urea | 2:1 |
100 s | 85 | NR | 16 |
| ZnO·La2CO5·LaOOH | Passiflora edulis oil | 2.3 | 100 | MEA | 3:1 |
8 h | 100 | 2 | 20 |
| Sodium methoxidea | Soybean oil | 1 | 60 | NME | 3:1 |
NR | 90 | NR | 30 |
| Sodium methylateap | Methyl laurate | 0.33 | 149 | DEA | 1.1:1 |
10 s | 91 | NR | 31 |
| Sodium ethoxidea | Palm oil | 59 | 78 | Urea | 6.2:1 |
8 h | 79 | NR | 32 |
| Na/Ca(OH)2 | Methyl laurate | 3.5 | 110 | DEA | 6:1 |
1 h | >99 | 6 | 41 |
| Zn dustc | Methyl benzoate | 0.5 | 70 | Aniline | 1:1 |
16 h | 82 | 6 | 46 |
| CaO–BB | Soybean oil | 20 | 130 | DEA | 1:2 |
3 h | 90.6 | NR | 47 |
| CaO | Soybean oil | 20 | 130 | MEA | 1:2 |
3 h | 80.6 | NR | 47 |
| K2CO3 | Soybean oil | 20 | 130 | MEA | 1:1 |
3 h | 72.7 | NR | 47 |
| KOHa | Soybean oil | 20 | 130 | MEA | 1:1 |
3 h | 74.9 | NR | 47 |
| Tetra-n-butyl titanate | Oleic acid | 0.25 | 135 | Urea | 1:5 |
25 min | 95 | NR | 48 |
| Ni/CaO | WO, SO, CO, KO, JO, MF/methyl laurate | 5 | 110 | DEA | 5:1 |
0.5 h | >99 | 7 | PR |
4. Conclusions
In the present work, Ni/CaO catalysts were prepared in nanocrystalline form and successfully employed for the aminolysis as well as transesterification reactions of a variety of triglycerides. The catalyst activity was found to be a function of its basic strength which depends upon the Ni content and calcination temperature. The catalyst was found to be effective even at room temperature (35 °C) and hence, the Ni/CaO catalyzed reaction doesn’t demand energy intensive reaction conditions to achieve product formation. The kinetic experiment suggests that Ni/CaO catalyzed aminolysis of triglycerides follows the pseudo first order rate law. The hot filtration test rules out any homogeneous contribution in catalytic activity. The catalyst was also recovered and successfully reused during 7 consecutive reaction cycles.Acknowledgements
We acknowledge DST for the financial support (Ref No. EMR/2014/000090). We are also thankful to SAIF (Panjab University, Chandigarh, India) for NMR and TEM, and SAI Lab (Thapar University, Patiala, India) for SEM studies.References
- B. Subhasree, R. Baskar, R. L. Keerthana, R. L. Susan and P. Rajasekaran, Food Chem., 2009, 115, 1213–1220 CrossRef CAS.
- R. Luque, L. Herrero-Davila, J. M. Campelo, J. H. Clark, J. M. Hidalgo, D. Luna, J. M. Marinas and A. A. Romero, Energy Environ. Sci., 2008, 1, 542–564 CAS.
- U. Biermann, W. Friedt, S. Lang, W. Lühs, G. Machmüller, J. O. Metzger, M. R. Gen Klaas, H. J. Schaefer and M. P. Schneider, Angew. Chem., Int. Ed., 2000, 39, 2206–2224 CrossRef CAS.
- H. Maag, J. Am. Oil Chem. Soc., 1984, 61, 259–267 CrossRef CAS.
- T. M. Kuo, H. Kim and C. T. Hou, Curr. Microbiol., 2001, 43, 198–203 CrossRef CAS PubMed.
- R. W. Johnson and E. Fritz, Fatty Acids in Industry: Processes, Properties, Derivatives, Applications, Marcel Dekker, New York and Basel, 1989 Search PubMed.
- S. Stournas, E. Lois and A. Serdari, J. Am. Oil Chem. Soc., 1995, 72, 433–437 CrossRef CAS.
- R. Alcantara, J. Amores, L. t. Canoira, E. Fidalgo, M. Franco and A. Navarro, Biomass Bioenergy, 2000, 18, 515–527 CrossRef CAS.
- P. D. Bailey, I. D. Collier and K. M. Morgan, in Comp. Org. Funct. Group, Transform, ed. A. R. K. M. C. W. Rees, Elsevier Science, Oxford, 1995 Search PubMed.
- C. G. de Almeida, G. D. Garbois, L. M. Amaral, C. C. Diniz and M. Le Hyaric, Biomed. Pharmacother., 2010, 64, 287–290 CrossRef CAS PubMed.
- N. D. Karis, W. A. Loughlin and I. D. Jenkins, Tetrahedron, 2007, 63, 12303–12309 CrossRef CAS.
- K. C. Nadimpally, K. Thalluri, N. B. Palakurthy, A. Saha and B. Mandal, Tetrahedron Lett., 2011, 52, 2579–2582 CrossRef CAS.
- M. Nakanishi, J.-S. Park, W.-D. Jang, M. Oba and K. Kataoka, React. Funct. Polym., 2007, 67, 1361–1372 CrossRef CAS.
- S. S. Bhella, M. Elango and M. P. S. Ishar, Tetrahedron, 2009, 65, 240–246 CrossRef CAS.
- C. Ferroud, M. Godart, S. Ung, H. Borderies and A. Guy, Tetrahedron Lett., 2008, 49, 3004–3008 CrossRef CAS.
- A. Khalafi-Nezhad, B. Mokhtari and M. N. Soltani Rad, Tetrahedron Lett., 2003, 44, 7325–7328 CrossRef CAS.
- W. E. Levinson, T. M. Kuo and G. Knothe, Bioresour. Technol., 2008, 99, 2706–2709 CrossRef CAS PubMed.
- L. Perreux, A. Loupy and F. Volatron, Tetrahedron, 2002, 58, 2155–2162 CrossRef CAS.
- S. van Pelt, R. L. M. Teeuwen, M. H. A. Janssen, R. A. Sheldon, P. J. Dunn, R. M. Howard, R. Kumar, I. Martinez and J. W. Wong, Green Chem., 2011, 13, 1791–1798 RSC.
- C. G. de Almeida, I. F. de Souza, R. A. Sousa and M. Le Hyaric, Catal. Commun., 2013, 42, 25–29 CrossRef CAS.
- M. C. de Zoete, A. C. Kock-van Dalen, F. van Rantwijk and R. A. Sheldon, J. Mol. Catal. B: Enzym., 1996, 1, 109–113 CrossRef CAS.
- W. E. Levinson, T. M. Kuo and C. P. Kurtzman, Enzyme Microb. Technol., 2005, 37, 126–130 CrossRef CAS.
- M. J. J. Litjens, M. Sha, A. J. J. Straathof, J. A. Jongejan and J. J. Heijnen, Biotechnol. Bioeng., 1999, 65, 347–356 CrossRef CAS PubMed.
- N. P. Awasthi and R. P. Singh, Eur. J. Lipid Sci. Technol., 2009, 111, 202–206 CrossRef CAS.
- B. C. Ranu and P. Dutta, Synth. Commun., 2003, 33, 297–301 CrossRef CAS.
- C. Han, J. P. Lee, E. Lobkovsky and J. A. Porco, J. Am. Chem. Soc., 2005, 127, 10039–10044 CrossRef CAS PubMed.
- V. M. de Oliveira, R. Silva de Jesus, A. F. Gomes, F. C. Gozzo, A. P. Umpierre, P. A. Z. Suarez, J. C. Rubim and B. A. D. Neto, ChemCatChem, 2011, 3, 1911–1920 CrossRef CAS.
- D. J. C. Constable, P. J. Dunn, J. D. Hayler, G. R. Humphrey, J. J. L. Leazer, R. J. Linderman, K. Lorenz, J. Manley, B. A. Pearlman, A. Wells, A. Zaks and T. Y. Zhang, Green Chem., 2007, 9, 411–420 RSC.
- P. Xiao, S. Zhang, H. Ma, A. Zhang, X. Lv and L. Zheng, J. Biotechnol., 2013, 168, 552–559 CrossRef CAS PubMed.
- J. W. Rawlins, M. Pramanik and S. K. Mendon, J. Am. Oil Chem. Soc., 2008, 85, 783–789 CrossRef CAS.
- J. Monick, J. Am. Oil Chem. Soc., 1962, 39, 213–215 CrossRef CAS.
- E. A. J. Al-Mulla, W. M. Z. W. Yunus, N. A. B. Ibrahim and M. Z. A. Rahman, J. Oleo Sci., 2009, 58, 467–471 CrossRef CAS PubMed.
- P. Mu and D. T. Plummer, Introduction to practical biochemistry, Tata McGraw-Hill Education, 1988, pp. 195–197 Search PubMed.
- T. Yamanaka and K. Tanabe, J. Phys. Chem., 1975, 79, 2409–2411 CrossRef CAS.
- D. Kumar and A. Ali, Energy Fuels, 2013, 27, 3758–3768 CrossRef CAS.
- X.-R. Chen, Y.-H. Ju and C.-Y. Mou, J. Phys. Chem. C, 2007, 111, 18731–18737 CAS.
- T. Horikawa, D. Do and D. Nicholson, Adv. Colloid Interface Sci., 2011, 169, 40–58 CrossRef CAS PubMed.
- V. Rives and S. Kannan, J. Mater. Chem., 2000, 10, 489–495 RSC.
- A. Bensalem, F. Bozon-Verduraz, M. Delamar and G. Bugli, Appl. Catal., A, 1995, 121, 81–93 CrossRef CAS.
- A. Bensalem, J. Muller and F. Bozon-Verduraz, J. Chem. Soc., Faraday Trans., 1992, 88, 153–154 RSC.
- D. Kumar, S. M. Kim and A. Ali, New J. Chem., 2015, 39, 7097–7104 RSC.
- D. Kumar and A. Ali, Fuel, 2015, 159, 845–853 CrossRef CAS.
- J. M. Balbino, E. W. de Menezes, E. V. Benvenutti, R. Cataluña, G. Ebeling and J. Dupont, Green Chem., 2011, 13, 3111–3116 RSC.
- N. Kaur and A. Ali, RSC Adv., 2014, 4, 43671–43681 RSC.
- T. Maugard, M. Remaud-Simeon, D. Petre and P. Monsan, Tetrahedron, 1997, 53, 5185–5194 CrossRef CAS.
- R. Arora, S. Paul and R. Gupta, Can. J. Chem., 2005, 83, 1137–1140 CrossRef CAS.
- J. Zhang, D. Cai, S. Wang, Y. Tang, Z. Zhang, Y. Liu and X. Gao, Can. J. Chem. Eng., 2014, 92, 871–875 CrossRef CAS.
- S. S. Hoong, S. Ahmad and H. A. Hassan, US Pat., 7098351, 2006.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra12114d |
| This journal is © The Royal Society of Chemistry 2016 |