DOI:
10.1039/C6RA12112H
(Paper)
RSC Adv., 2016,
6, 78984-78993
Synthesis and characterization of triphenylamine-based polymers and their application towards solid-state electrochromic cells†
Received
10th May 2016
, Accepted 3rd August 2016
First published on 15th August 2016
Abstract
Four novel triphenylamine-based polymers, PJK1, PJK2, PJK3 and PJK4 were successfully synthesized and fully characterized by 1H NMR, UV-VIS spectroscopy, cyclic voltammetry (CV), GPC and spectroelectrochemistry. These polymers are easily soluble in many common organic solvents, which make them appropriate for film deposition via spray-coating. We fabricated electrochromic cells comprising ITO-coated glass/polymer/gel electrolyte/ITO-coated glass and patterned the color change by applying direct current with different voltages. We report herein color changes, from the neutral to oxidized form as follows: for PJK1, orange to dark green; for PJK2, light yellow to reddish brown; for PJK3, light blue to grey; and for PJK4, green to bluish green. The majority of the copolymers exhibited very good thermal stabilities, as evidenced by less than a 5% weight loss in temperatures exceeding 400 °C. To further characterize, we simulated the electrochemical and optical properties, which are good agreement with the experimental data.
1. Introduction
Electrochromism is generally known as a reversible optical change in a material induced by an external voltage; many organic and inorganic compounds exhibit electrochromism throughout the electromagnetic spectrum.1 The idea of an electrochromic display was first suggested in 1961.2 This property led to the expansion of many technological applications such as smart windows,3 automatic anti-glazing mirrors,4 chameleon materials,5 and electrochromic displays.6,7 In recent years, WO3 (ref. 4) and viologen-based8 electrochromic devices were commercialized. In the series of electrochromic compounds, conjugated polymers can display high coloration over a wide color range at low operating voltage with fast switching capabilities, high optical contrasts, rapid response times and the ability to modify their structure to create multicolor electrochromes.9 Conjugated electrochromic polymers, such as poly(thiophene) (PTh), poly(aniline) (PANI) and poly(pyrrole) (PPy), have been extensively studied.10 Previously, many electrochromic organic polymers were obtained by electropolymerization or oxidative polymerization,11,12 but chemical polymerization has more advantages than these polymerization methods. Because in electropolymerization or oxidative polymerization methods we always get the problems, such as the lack of film uniformity over large surface, low material recovery, inability to form large amounts of processable polymer and irregular linkages within the polymer backbone.13,14
Triphenylamine (TPA) derivatives, which are electron-donating in nature, show excellent electrochemical stability, optoelectronic and electrochromic behavior.15–18 The characteristic structural feature of triphenylamine is the nitrogen center, which also serves as its electroactive site. The nitrogen atom is connected to three electron-rich phenyl groups in a propeller-like geometry.19 Electron-rich triphenylamine derivatives are easily oxidized to form stable polarons, an oxidation process that is always associated with a clear change in coloration of the molecule. Additionally, different thiophene-based derivatives have gained much attention during the past years, owing to the variation of the optical and electronic properties by simple structural modifications.20,21 Fluorene is a biphenyl structure with rigid plane and huge π-conjugated molecule, whose derivatives have been widely used in organic electro-luminescence materials, photochromic materials two-photon absorbing materials, because of their very high photoluminescence, thermal stability, electroluminescence quantum efficiency, good solubility.22–24
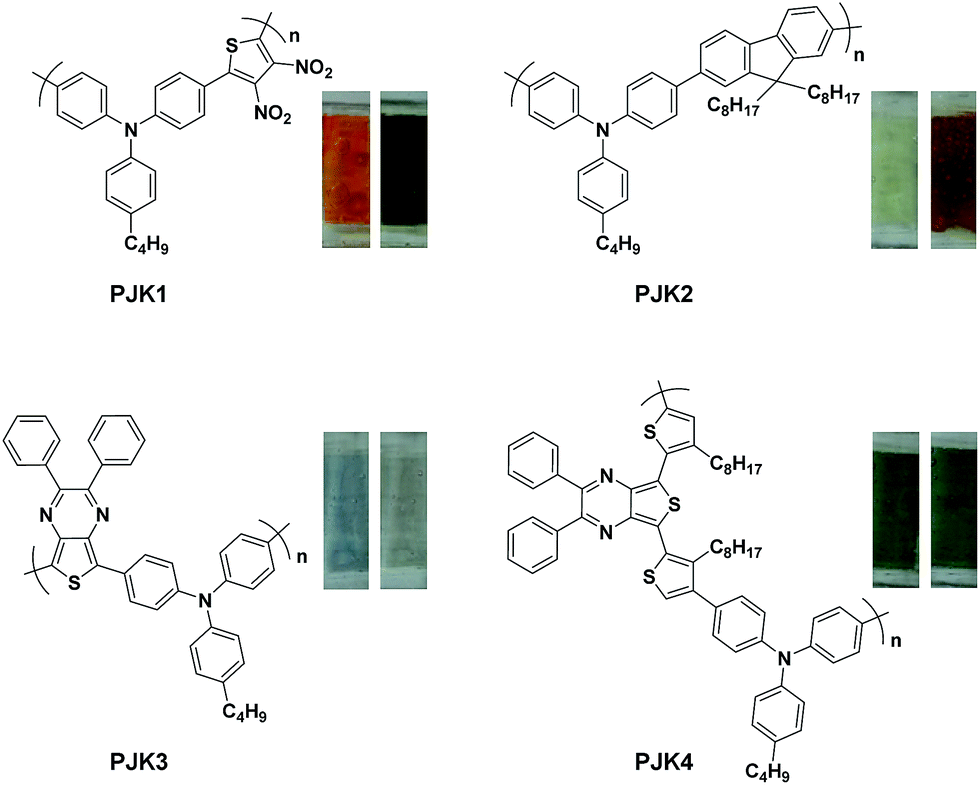
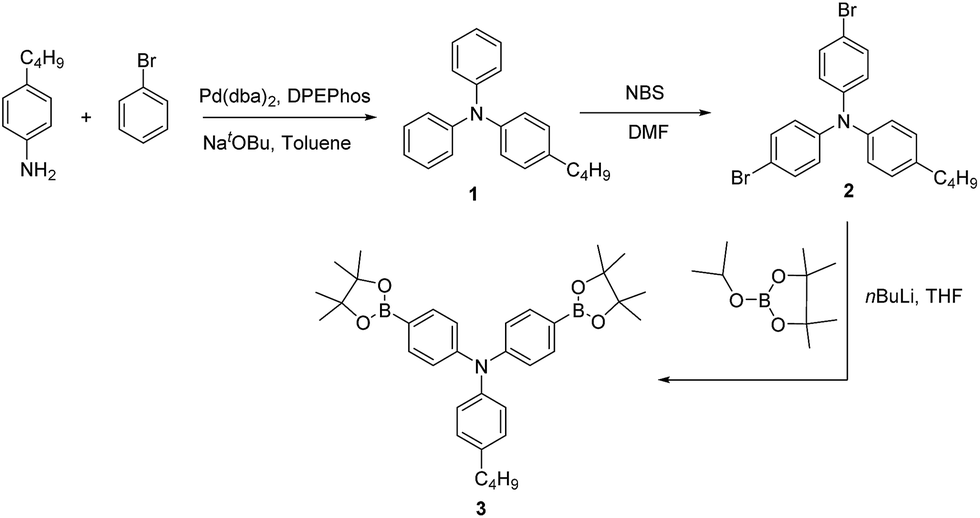
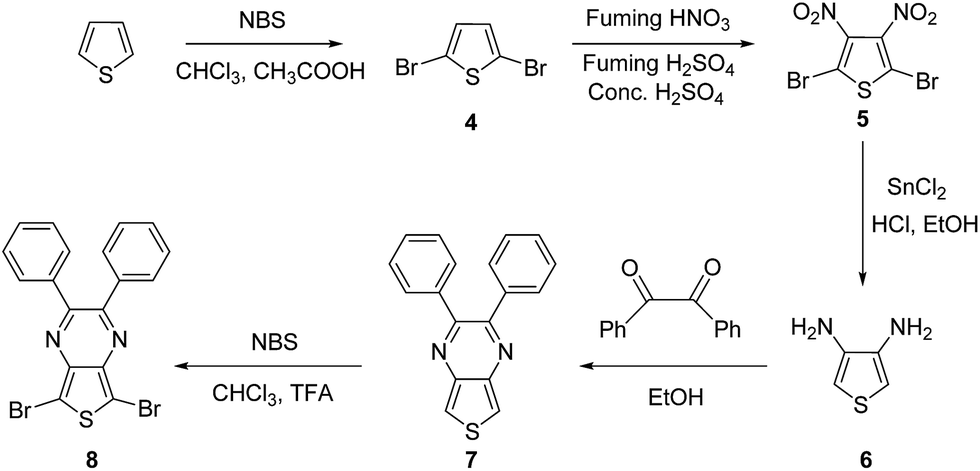
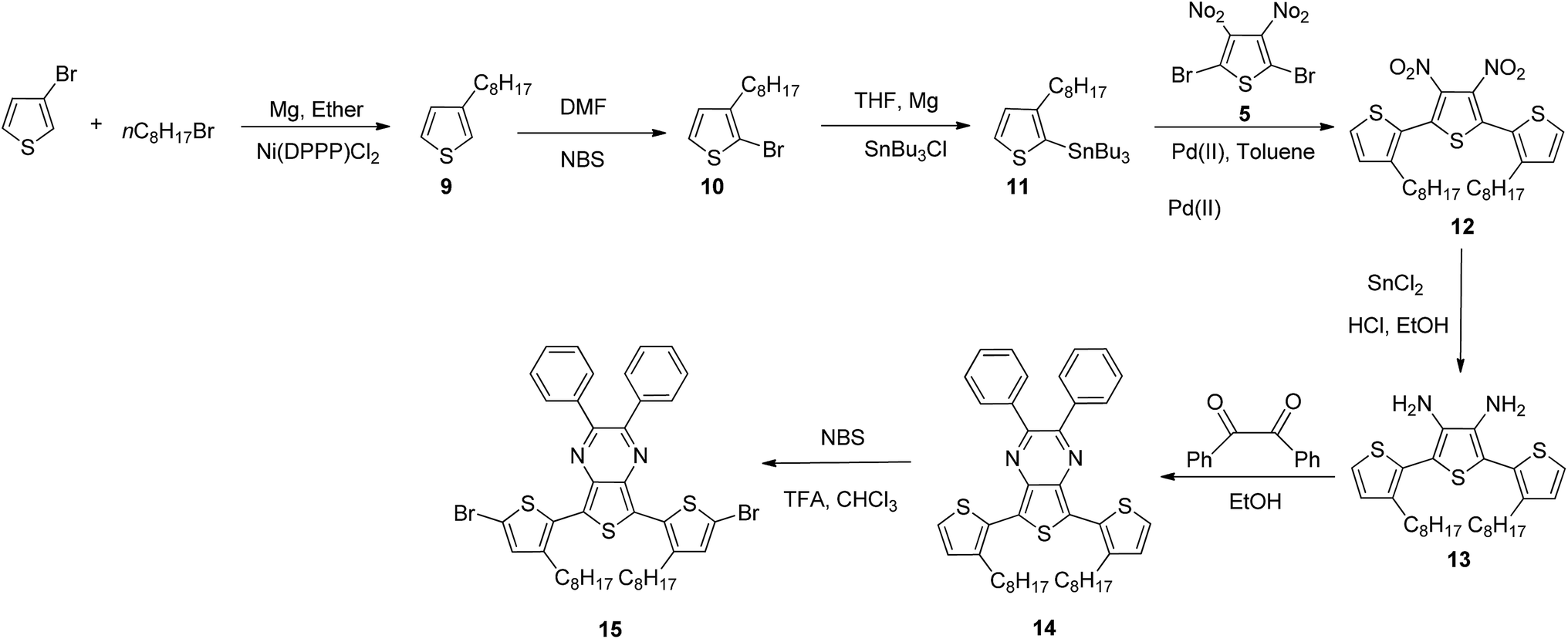
Here in this research we designed a series of conjugated polymers as EC cells, wherein, triphenylamine (TPA) and thiophene or fluorene were chosen to build the backbone of our target molecule with the principle of different color properties and these copolymers are highly conjugated and giving a small band gap should therefore be more conducting nature. We reported the synthesis and full characterization of four triphenylamine-based polymers: PJK1, PJK2, PJK3 and PJK4. The resulting copolymers were readily soluble in most industrial solvents such as chloroform, dichloromethane, DMF, and tetrahydrofuran. To evaluate the electrochemical properties, all-solid-state electrochemical cells were prepared by spray-coating chloroform solutions of the copolymers onto ITO-coated glass and used a polymer gel as the electrolyte. Neutral-to-oxidized color changes for the copolymers all differed, exhibiting transitions from orange to dark green, light yellow to reddish brown, light blue to grey, and green to bluish green (Fig. 1).
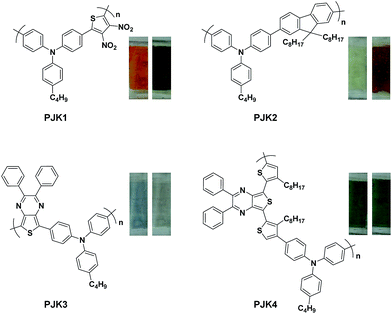 |
| | Fig. 1 Polymer structures and color transitions. | |
2. Experimental section
2.1. Materials and characterization
All solvents and reagents (analytical and spectroscopic grades) were commercially obtained and used as received unless otherwise noted. Fourier 300, 7.05 tesla, 300 MHz, an AVANCE III 600 spectrometer (Akishima, Japan) was operated at 600 MHz and 150 MHz for 1H and 13C NMR spectroscopy, respectively. CDCl3 was used as the solvent, and Alice 4.0 software was used for analysis. The chemical shifts (δ values) are reported in ppm downfield from an internal standard (Me4Si). Mass spectra were recorded on a 4000 Q TRAP mass spectrometer. HRMS spectra were obtained using a micrOTOF-QII mass spectrometer. FT-IR spectra were recorded on an ALPHA-P spectrometer. UV-visible absorption spectra were recorded using an Agilent 8453 spectrophotometer. The electrochemical measurements, i.e., cyclic voltammetry (CV) experiments, were conducted using a Versa STAT 3 instrument using polymer films sprayed on platinum button, the electrolyte used was 0.1 M TBABF4 in propylene carbonate, Ag/Ag+ electrode as the reference electrode and platinum wire as the counter electrode.
Thermogravimetric analyses (TGA) were performed on a Mettler Toledo instrument using a heating rate of 20 °C min−1 and nitrogen flow rate of 50 mL min−1. Number-average (Mn) and weight-average (Mw) molecular weights were determined by gel permeation chromatography (GPC) using a Waters model 2690 instrument with THF (HPLC grade, Aldrich) as the eluting solvent. Spectroelectrochemical data of the polymers films were recorded by using an Instek model GPS-3303 instrument as a DC power source. Electrochromic polymer films were produced by spraying solutions of the polymers (5 mg mL−1 in chloroform) onto indium tin oxide (ITO)-coated glass slides (square: surface resistivity: 8–12 Ω sq−1, 25 mm × 25 mm × 1.1 mm, Sigma-Aldrich). The polymers were spray-coated onto the active surface of an ITO plate using a mask.
2.2. Synthesis of monomer
2.2.1. 4-Butyl-N,N-diphenylaniline (1). Bromobenzene (26.69 g, 170 mmol), 4-butylaniline (8.344 g, 56 mmol) and sodium tert-butoxide (19.23 g, 200 mmol) were added to a solution of bis[2-(diphenylphosphino)phenyl]ether (DPEPhos) (0.914 g, 1.7 mmol) and bis(dibenzylideneaceton)palladium (Pd(dba)2) (0.789 g, 1.3 mmol) in toluene (160 mL) under an inert atmosphere. The resulting solution was stirred at 90 °C for 24 h. The reaction mixture was allowed to cool to room temperature, at which point 500 mL water was added, and the mixture was extracted with ethyl acetate (2 × 250 mL). The combined organic layers were dried over sodium sulfate, concentrated, and purified by silica gel column chromatography (10% ethyl acetate in hexane) to yield 14.02 g (83%) of the product as a yellow and viscous oil. 1H NMR (300 MHz, CDCl3): δ 7.17–7.08 (m, 4H), 7.01–6.83 (m, 10H), 2.48 (t, J = 7.7 Hz, 2H), 1.57–1.44 (m, 2H), 1.35–1.21 (m, 2H), 0.85 (t, J = 7.3 Hz, 3H) ppm; 13C NMR (75 MHz, CDCl3): δ 148.1, 145.3, 137.7, 129.2, 129.1, 124.7, 123.7, 122.1, 35.0, 33.6, 22.4, 13.9 ppm; IR νmax in cm−1 (in CHCl3): 2930, 1589, 1508, 1491, 1276, 1075, 903, 833, 723, 649, 621; ESI-MS: 302 [M + H]+.
2.2.2. 2,3-Diphenylthieno[3,4-b]pyrazine (7). Compound 6 (0.45 g, 3.95 mmol) was added to benzil (0.91 g, 4.34 mmol) in ethanol (85 mL) and refluxed overnight. The solution was evaporated and purified by column chromatography (10% ethyl acetate in hexane) to afford a yellow solid (0.81 g, 71%). 1H NMR (600 MHz, CDCl3): δ 8.05 (s, 2H), 7.46–7.29 (m, 10H) ppm; 13C NMR (150 MHz, CDCl3): δ 153.4, 141.6, 139.1, 129.6, 128.8, 128.1, 117.5 ppm; IR νmax in cm−1 (KBr): 3084, 3054, 1597, 1576, 1231, 1074, 839, 816, 723; ESI-MS: 289 [M + H]+.
2.2.3. 5,7-Dibromo-2,3-diphenylthieno[3,4-b]pyrazine (8). NBS (0.97 g, 5.4 mmol) was added to compound 7 (0.75 g, 2.6 mmol) in a mixture of chloroform/trifluoroacetic acid (16 mL; 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v), and the mixture was stirred overnight in the dark. The mixture was extracted with ethyl acetate and dried over sodium sulfate, evaporated, and purified by column chromatography (50% dichloromethane in hexane) to afford a greenish-yellow solid (0.86 g, 74%). 1H NMR (300 MHz, CDCl3): δ 7.46–7.27 (m, 10H) ppm; 13C NMR (150 MHz, CDCl3): δ 154.7, 139.3, 138.2, 129.8, 129.3, 128.1, 104.9 ppm; IR νmax in cm−1 (KBr): 3051, 1596, 1523, 1253, 1025, 811, 763, 739; HR-MS (ESI-MS) m/z calcd for C18H11Br2N2S [M + H]+: 446.8984, found 446.8989.
:1 v/v), and the mixture was stirred overnight in the dark. The mixture was extracted with ethyl acetate and dried over sodium sulfate, evaporated, and purified by column chromatography (50% dichloromethane in hexane) to afford a greenish-yellow solid (0.86 g, 74%). 1H NMR (300 MHz, CDCl3): δ 7.46–7.27 (m, 10H) ppm; 13C NMR (150 MHz, CDCl3): δ 154.7, 139.3, 138.2, 129.8, 129.3, 128.1, 104.9 ppm; IR νmax in cm−1 (KBr): 3051, 1596, 1523, 1253, 1025, 811, 763, 739; HR-MS (ESI-MS) m/z calcd for C18H11Br2N2S [M + H]+: 446.8984, found 446.8989.
2.2.4. 3,3′′-Dioctyl-[2,2′:5′,2′′-terthiophene]-3′,4′-diamine (13). Compound 12 (3.03 g, 5.4 mmol) was stirred in a mixture of HCl/ethanol (24 mL/35 mL) and cooled in an ice bath. Tin(II) chloride dihydrate (40 g, 177 mmol) was added in small portions to maintain the temperature at 25 °C. After the reaction was stirred overnight, the pH was adjusted to 10 with 4 M KOH, and the solution was extracted with ethyl acetate. The organic layer was dried with sodium sulfate and purified by column chromatography (70% ethyl acetate in hexane) to afford the desired compound as a brown oil (1.96 g, 72%). 1H NMR (300 MHz, CDCl3): δ 7.29 (d, J = 5.3 Hz, 2H), 6.99 (d, J = 5.1 Hz, 2H), 3.35 (s, 4H), 2.62 (t, J = 7.7 Hz, 4H), 1.65–1.52 (m, 4H), 1.37–1.15 (m, 20H), 0.87 (t, J = 6.5, 6H) ppm; 13C NMR (150 MHz, CDCl3): δ 142.33, 134.57, 129.28, 127.88, 125.29, 109.01, 31.88, 30.81, 29.45, 29.39, 29.25, 28.94, 22.65, 14.08 ppm; IR νmax in cm−1 (in CHCl3): 3413, 3325, 2922, 2852, 1613, 1572, 1496, 1451, 904, 829, 725, 648; ESI-MS: 503 [M + H]+.
2.2.5. 5,7-Bis(3-octylthiophen-2-yl)-2,3-diphenylthieno[3,4-b]pyrazine (14). Compound 13 (1.85 g, 3.7 mmol) was added to a solution of benzil (0.85 g, 4.0 mmol) in ethanol (65 mL) and refluxed overnight. The solution was concentrated by evaporation and purified by column chromatography (10% ethyl acetate in hexane) to afford the desired compound as a purple solid (2.32 g, 92%). 1H NMR (300 MHz, CDCl3): δ 7.55–7.42 (m, 4H), 7.41–7.29 (m, 8H), 7.03 (d, J = 5.1 Hz, 2H), 2.97 (t, J = 7.7 Hz, 4H), 1.81–1.68 (m, 4H), 1.43–1.14 (m, 20H), 0.86 (t, J = 6.5 Hz, 6H) ppm; 13C NMR (150 MHz, CDCl3): δ 152.3, 141.0, 139.1, 137.7, 134.8, 130.0, 129.5, 128.8, 127.9, 126.8, 125.3, 31.9, 30.4, 30.3, 29.7, 29.5, 29.3, 22.7, 14.1 ppm; IR νmax in cm−1 (KBr): 3083, 3060, 2951, 1577, 1511, 1246, 1209, 713, 694, 640; ESI-MS: 678 [M + H]+.
2.2.6. 5,7-Bis(5-bromo-3-octylthiophen-2-yl)-2,3-diphenyl thieno[3,4-b]pyrazine (15). NBS (1.14 g, 6.4 mmol) was added to a solution of compound 14 (1.97 g, 2.9 mmol) in a mixture of chloroform/trifluoro acetic acid (14 mL; 1:1 v/v), and the mixture was stirred overnight in dark. The reaction mixture was extracted with ethyl acetate, dried over sodium sulfate and purified by column chromatography (50% dichloromethane in hexane) to afford the desired compound as a greenish, dark purple solid (2.39 g, 98%). 1H NMR (300 MHz, CDCl3): δ 7.55 (d, J = 6.2 Hz, 4H), 7.39–7.30 (m, 6H), 6.98 (s, 2H), 2.91 (t, J = 7.7 Hz, 4H), 1.78–1.66 (m, 4H), 1.45–1.18 (m, 20H), 0.87 (t, J = 6.7 Hz, 6H) ppm; 13C NMR (150 MHz, CDCl3): δ 152.79, 141.14, 138.80, 137.65, 132.14, 130.07, 129.62, 129.06, 128.10, 124.24, 114.44, 31.87, 30.53, 30.06, 30.08, 29.73, 29.49, 29.29, 22.66, 14.08 ppm; IR νmax in cm−1 (KBr): 3019, 1215, 905, 753, 726; HR-MS (ESI-MS) m/z calcd for C42H46Br2N2S3Na [M + Na]+: 857.1064, found 857.1037.
2.3. Copolymer synthesis
2.3.1. Poly(4-butyl-N-(4-(3,4-dinitrothiophen-2-yl)phenyl)-N-phenylaniline) (PJK1). 4-Butyl-N,N-bis(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)aniline (0.2 g, 0.37 mmol), 2,5-dibromo-3,4-dinitrothiophene (0.12 g, 0.37 mmol), Pd(0)(PPh3)4 (0.02 g, 0.017 mmol), and aqueous 2 M K2CO3 (5 mL) were added to dry THF (15 mL). The mixture was vigorously stirred at 85–90 °C for 48–72 h. After the solution cooled to room temperature, the reaction mixture was poured into a cold mixture of methanol/deionized water (10:1 v/v). The solid polymer was collected by filtration and washed with methanol. The recovered polymer was then washed for 24 h in a Soxhlet apparatus using acetone to remove oligomers and catalyst residues. Finally, the polymer was redissolved in chloroform, precipitated from cold methanol and dried (112 mg, 61%). 1H NMR (600 MHz, CDCl3): δ 7.42–7.38 (m, 4H), 7.22–7.09 (m, 8H), 2.63 (t, J = 7.5, 2H), 1.66–1.60 (m, 2H), 1.43–1.35 (m, 2H), 0.96 (t, J = 7.24, 3H) ppm.
2.3.2. Poly(4-butyl-N-(4-(9,9-dioctyl-9H-fluoren-2-yl)phenyl)-N-phenylaniline) (PJK2). The procedure used to synthesize PJK1 was followed, using 4-butyl-N,N-bis(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)aniline (0.2 g, 0.37 mmol), 2,7-dibromo-9,9-dioctyl-9H-fluorene (0.2 g, 0.37 mmol), Pd(0)(PPh3)4 (0.02 g, 0.017 mmol), THF (15 mL) and 2 M K2CO3 (5 mL). Yield (140 mg, 56%). 1H NMR (600 MHz, CDCl3): δ 7.76–7.72 (m, 2H), 7.60–7.54 (m, 8H), 7.24–7.19 (m, 4H), 7.15–7.11 (m, 4H), 2.64–2.59 (m, 2H), 2.60–1.92 (br, 4H), 1.66–1.60 (m, 2H), 1.43–1.37 (m, 2H), 1.31–1.02 (m, 16H), 0.99–0.94 (t, J = 7.34, 4H), 0.83–0.77 (m, 9H) ppm.
2.3.3. Poly(4-butyl-N-(4-(2,3-diphenylthieno[3,4-b]pyrazin-5-yl)phenyl)-N-phenylaniline) (PJK3). The procedure used to synthesize PJK1 was followed, using 4-butyl-N,N-bis(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)aniline (0.2 g, 0.37 mmol), 5,7-dibromo-2,3-diphenylthieno[3,4-b]pyrazine (0.17 g, 0.37 mmol), Pd(0)(PPh3)4 (0.02 g, 0.017 mmol), THF (15 mL) and 2 M K2CO3 (5 mL). Yield (108 mg, 51%). 1H NMR (600 MHz, CDCl3): δ 8.23–8.17 (m, 4H), 7.55–7.50 (m, 4H), 7.36–7.27 (m, 6H), 7.25–7.21 (m, 2H), 7.18–7.07 (m, 6H), 2.64–2.57 (m, 2H), 1.67–1.60 (m, 2H), 1.43–1.37 (m, 2H), 0.99 (t, J = 7.53, 3H) ppm.
2.3.4. Poly(4-butyl-N-(4-(4-octyl-5-(7-(3-octylthiophen-2-yl)-2,3-diphenylthieno[3,4-b]pyrazin-5-yl)thiophen-3-yl)phenyl)-N-phenylaniline) (PJK4). The procedure used to synthesize PJK1 was followed, using 4-butyl-N,N-bis(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)aniline (0.2 g, 0.37 mmol), 5,7-bis(5-bromo-3-octylthiophen-2-yl)-2,3-diphenylthieno[3,4-b]pyrazine (0.31 g, 0.37 mmol), Pd(0)(PPh3)4 (0.02 g, 0.017 mmol), THF (15 mL) and 2 M K2CO3 (5 mL). Yield (184 mg, 52%). 1H NMR (600 MHz, CDCl3): δ 7.63–7.53 (m, 8H), 7.37–7.29 (m, 6H), 7.21–7.09 (m, 10H), 3.03–2.97 (m, 4H), 2.64–2.58 (m, 2H), 1.84–1.16 (m, 28H), 0.99–0.79 (m, 9H) ppm.
3. Results and discussion
3.1. Monomer synthesis
The synthetic routes for all of the monomers are shown in Schemes 1–3. In Scheme 1, compound 1 was prepared by using the bis(dibenzylideneaceton)-palladium, ligand (bis-[2-(diphenylphosphino)phenyl]ether); compounds 2–6 were synthesized according to reported procedures.25–30 Compound 7 was successfully prepared by condensation with benzil in ethanol. In IR spectrum disappearance of the N–H stretching band of the amino functionality at 3355 cm−1 was indicated successful arylation to compound 7. Compound 8 was synthesized in good yield using NBS in a mixture of TFA and chloroform at room temperature (Scheme 2). The bromination was confirmed by disappearance of the peak at δ 8.05 in the 1H NMR spectrum and characterization data for compound 8 fully matched with the reported data (Scheme 2).31 Compound 9–12 prepared according to reported procedure.32–38 To synthesize compound 15 (Scheme 3), we modified a reported route.38 First, compound 12 was reduced using a mixture of HCl/ethanol and tin(II) chloride to compound 13. The previously reported yield for this reaction is 52%, which we were able to increase to 72% here. Subsequently, compound 13 was condensed with the benzil to give 14 in 92% yield, and then brominated with NBS in TFA and chloroform to afford compound 15 in 98% yield. Using this method, we obtained a higher overall yield for these 3 steps compared with that obtained using the previous method. The structures of all the synthesized compounds were confirmed by 1H NMR, 13C NMR, IR and mass spectral analyses. Compound 16 was obtained from a commercial source.
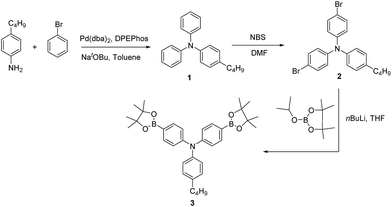 |
| | Scheme 1 Synthesis of 4-butyltriphenylamine derivative. | |
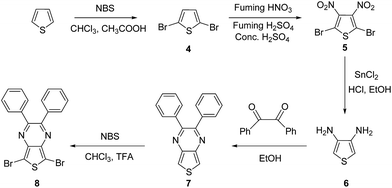 |
| | Scheme 2 Synthesis of 5,7-dibromo-2,3-diphenylthieno[3,4-b]pyrazine. | |
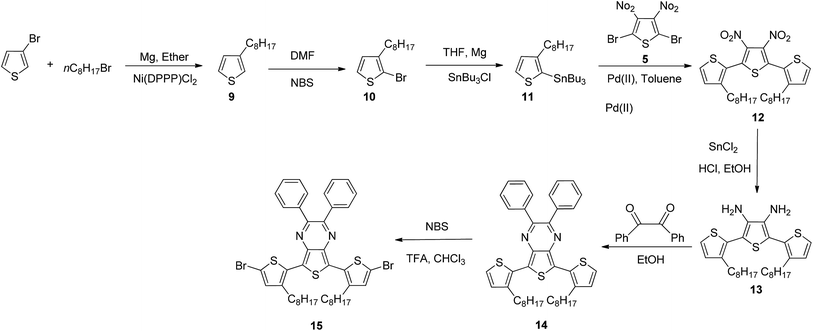 |
| | Scheme 3 Synthesis of 5,7-bis(5-bromo-3-octylthiophen-2-yl)-2,3-diphenylthieno[3,4-b]pyrazine. | |
3.2. Polymer synthesis
Polymers PJK1, PJK2, PJK3, and PJK4 were synthesized by the Suzuki coupling reaction of 4-butyl-N,N-bis(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)aniline (3) with compounds 5, 8, 15 and 16, respectively. The reactions were performed in the presence of Pd(PPh3)4 and potassium carbonate in THF solution (Scheme 4).39 The 1H NMR spectrum of PJK1 in CDCl3 showed peaks indicative of the 12 protons assignable to the phenyl rings of triphenylamine at δ 7.30–7.36 ppm and 6.98–7.16 ppm, while signals of the Ar-CH2 alkyl side chain of the phenyl units appeared at δ 2.56 ppm as a triplet, and the terminal –CH3 appeared as a triplet at δ 0.89. Similarly, the 1H NMR spectrum of PJK2 in CDCl3 exhibited peaks attributable to the 18 protons of the fluorene and phenyl groups in the aromatic region. The Ar-CH2 alkyl side chain on the phenyl units appeared at δ 2.56 ppm as a triplet, while the –CH3 protons appeared at δ 0.89 as a triplet. The 1H NMR spectrum of PJK3 in CDCl3 revealed peaks in the aromatic region for the phenyl groups, and the peaks in the aliphatic region was also confirmed the structure. For the spectrum of PJK4, the two protons of the thiophene–CH2 alkyl side chain appeared at δ 2.93 ppm as a broad signal, and protons attributable to the Ar-CH2 alkyl side chain on the phenyl units appeared at δ 2.53 ppm as a triplet. Detailed 1H NMR data of the polymers can be found in the Experimental section, while the 1H NMR spectra can be found in the ESI.†
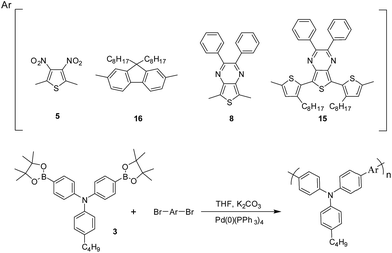 |
| | Scheme 4 Synthesis of copolymers. | |
3.3. GPC and TG analysis
The number-average molecular weight (Mn) and weight-average molecular weight (Mw) of the copolymers ranged from 2600 to 5400 and 4200 to 21600, respectively, with polydispersity index ranging from 1.6 to 4.0. These data are summarized in Table 1. The thermal properties of all the copolymers were investigated by TGA under a nitrogen atmosphere and data are summarized in Table 1. Most copolymers exhibited very good thermal stabilities, losing less than 5% of their weight until well above 400 °C. The amount of carbonized residue (char yield) of these polymers in nitrogen atmosphere was more than 61% at 600 °C.
Table 1 GPC, TGA and CV oxidation potential data of polymer derivatives
| Polymer |
Mn |
Mw |
PDI |
Tdec in °C |
Eox vs. SCE |
| PJK1 |
5400 |
12400 |
2.3 |
288 |
0.83 |
| PJK2 |
4700 |
15000 |
3.1 |
467 |
0.67 |
| PJK3 |
2600 |
4200 |
1.6 |
502 |
0.36 |
| PJK4 |
5300 |
21600 |
4.0 |
460 |
0.46 |
3.4. Cyclic voltammetry studies
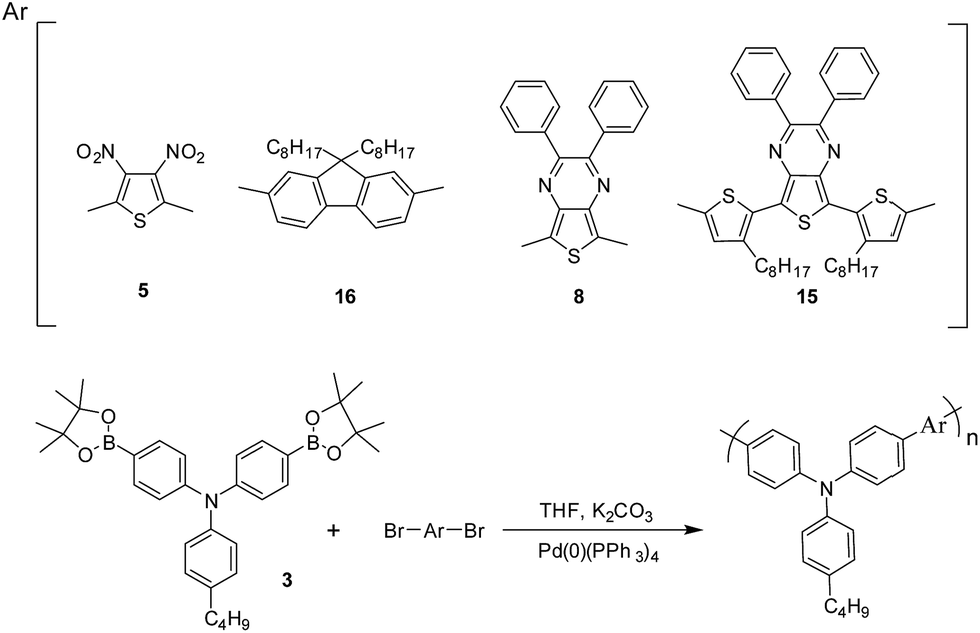
The cyclic voltammograms (CVs) of all the polymers can be found in Fig. 2 and the results are summarized in Table 1. Cyclic voltammograms were obtained by casting films of polymers onto a platinum button, propylene carbonate using 0.1 M TBABF4 as the supporting electrolyte, an Ag/Ag+ electrode as the reference electrode and a platinum wire as the counter electrode, at a scan rate of 50 mV s−1. The oxidation voltages of PJK1–PJK4 films were 0.83 V, 0.67 V, 0.36 V and 0.46 V respectively. All new polymers are stable upon oxidation with potential of oxidation ranging from 0.36 to 0.83 V vs. SCE. Here almost we observed that oxidation peaks has higher peak currents than reduction peaks and it shows these polymers better electron donors than acceptors.41
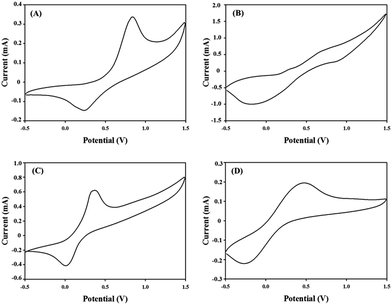 |
| | Fig. 2 Cyclic voltammograms of (A) PJK1; (B) PJK2; (C) PJK3; (D) PJK4 on platinum button with propylene carbonate in 0.1 M tetrabutylammonium tetrafluoro borate. Scan rate: 50 mV s−1 at 18 °C. | |
Cycling stability upon oxidation of all polymers has been conducted (see the ESI†). A thin film of polymer was placed on a platinum button and cyclic voltammetry was done at speed rate of 50 mV s−1. The cyclic voltammograms of PJK1 between −0.5 and +1.5 V vs. Ag/Ag+ shows a slight shift of the anodic peak (0.02 V) and a reduction of the current density of 40% after 500 cycles. For PJK2, the cyclic voltammograms at same conditions shows a shift of the anodic peak (+0.02 V) and a decrease in the current density after 500 cycles. For PJK3 current decreased around 30%. Finally PJK4 shows a shift of the anodic peak (+0.02 V) and a decrease of 30% in the current density. The minor shift of the anodic peak and the reduction of the current densities upon cycling can be attributed to an alteration in the film morphology or activation of new electro active site from freshly cast polymer film upon the repetitive insertion and extraction of counter ion through the oxidation process.38
3.5. Spectroelectrochemical studies
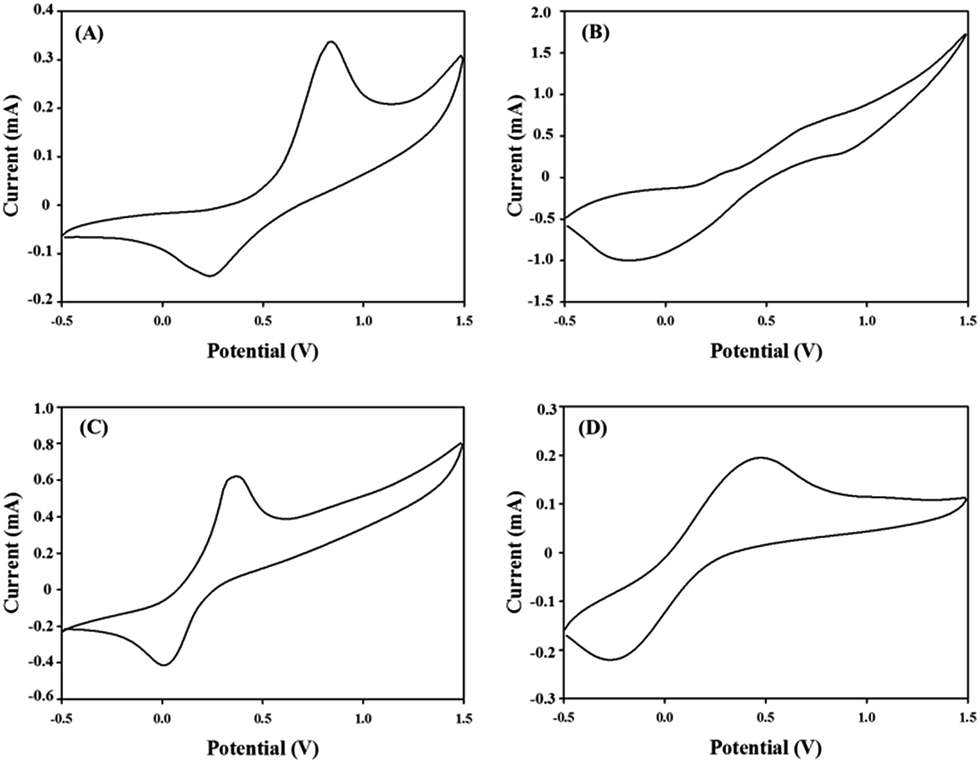
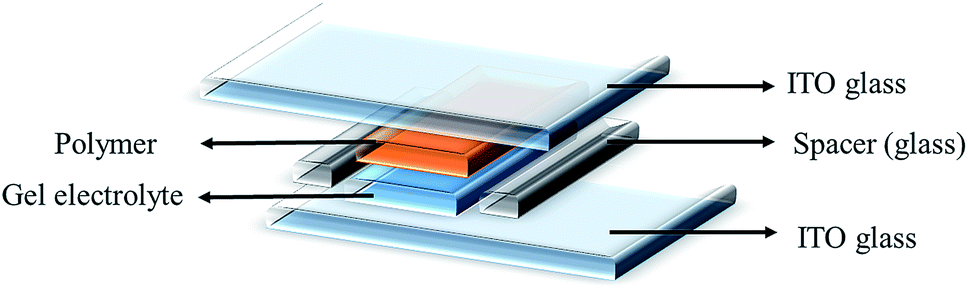
Spectroelectrochemical measurements were performed on films of the polymers deposited onto ITO-coated glass substrates following the spray-coating technique reported by Reynolds et al.,40 which facilitated formation of uniform polymeric layers. The gel electrolyte was spread on the polymer-coated side of the electrode, another ITO glass was sandwiched under atmospheric conditions, and glass spacers were used to fix the cell. To prevent leakage, an epoxy resin was applied to seal the device (see Fig. 3). The gel electrolyte prepared by using PMMA, LiClO4 and propylene carbonate in dry acetonitrile.39 This electrolyte allow ion transport and utilized to afford a single layer electrochromic cell (Fig. 4). DC voltage was fixed between 0.0 and 2.8 V for PJK1. At 0.0 V, polymer PJK1 is in a neutral state, and the color is orange. The change in absorption of the film of PJK1 at various applied potentials is shown in Fig. 5A. In the neutral form, at 0 V, PJK1 exhibited a strong absorption at 438 nm. Upon application of the voltage, this absorption band decreased while a new absorption peak starting at 580 nm and extending to above 900 nm in the NIR region grew in, while the color changed to green. The new absorption band exhibited a higher extinction coefficient value at 2 V, and then decreased after that voltage. The color change of the fabricated electrochromic cell was uniformed across the film.
 |
| | Fig. 3 Schematic cross-section of electrochromic cells. | |
 |
| | Fig. 4 Polymers with before and after applied potentials. | |
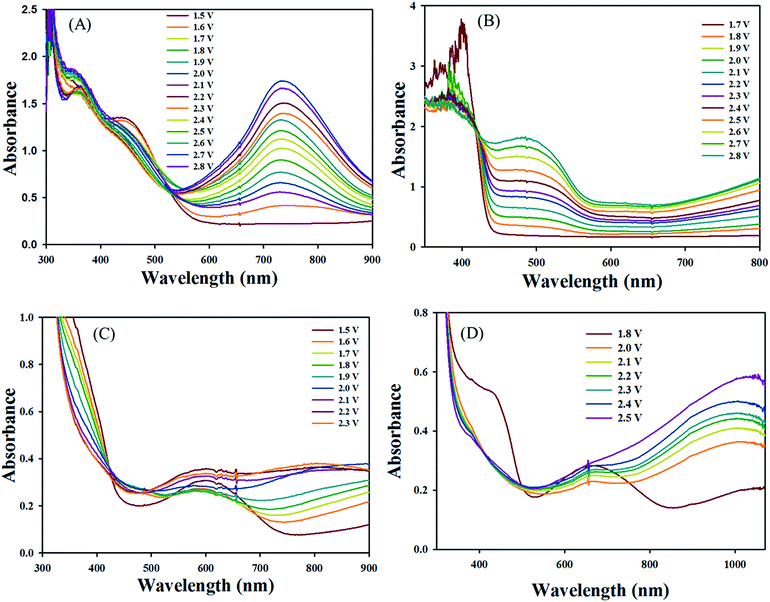 |
| | Fig. 5 Absorption spectra of PJK1 (A), PJK2 (B), PJK3 (C) and PJK4 (D) solid state EC using gel electrolyte (DC power supply). | |
Fig. 5B displays the spectroelectrochemical spectrum of PJK2 at various DC voltages up to 2.8 V. When a voltage of 0.0 V was applied, the polymer was in the neutral state with a light yellow color. When the applied voltage was increased to a maximum of 2.8 V, the color changed from light yellow to reddish brown with a uniform color change across the entire film. For potentials up to 1.7 V, PJK2 exhibited an absorption band at 399 nm; when the voltage was increased, this absorption band slowly diminished while a new absorption peak appeared between 430 and 580 nm, corresponding to a reddish brown color. PJK3, which was light blue in color, exhibited absorption maxima at 598 nm up to 1.5 V. By increasing the DC voltage, this absorption peak decreased, and a new absorption peak appeared in the region of 680 to 900 nm, resulting in a grey color (Fig. 5C). Finally, PJK4 exhibited a green color in its neutral state at 0.0 V, corresponding to an absorption maximum at 655 nm. Upon increasing the voltage, a new peak appeared starting at 700 nm and extending above 900 nm in the NIR region, resulting in a color change to bluish green (Fig. 5D). Moreover, these polymers displayed high molar extinction coefficient values in solution state, which was confirmed to present a good coloration efficiency (see ESI, Fig. 49†).42
3.6. Electrochromic switching
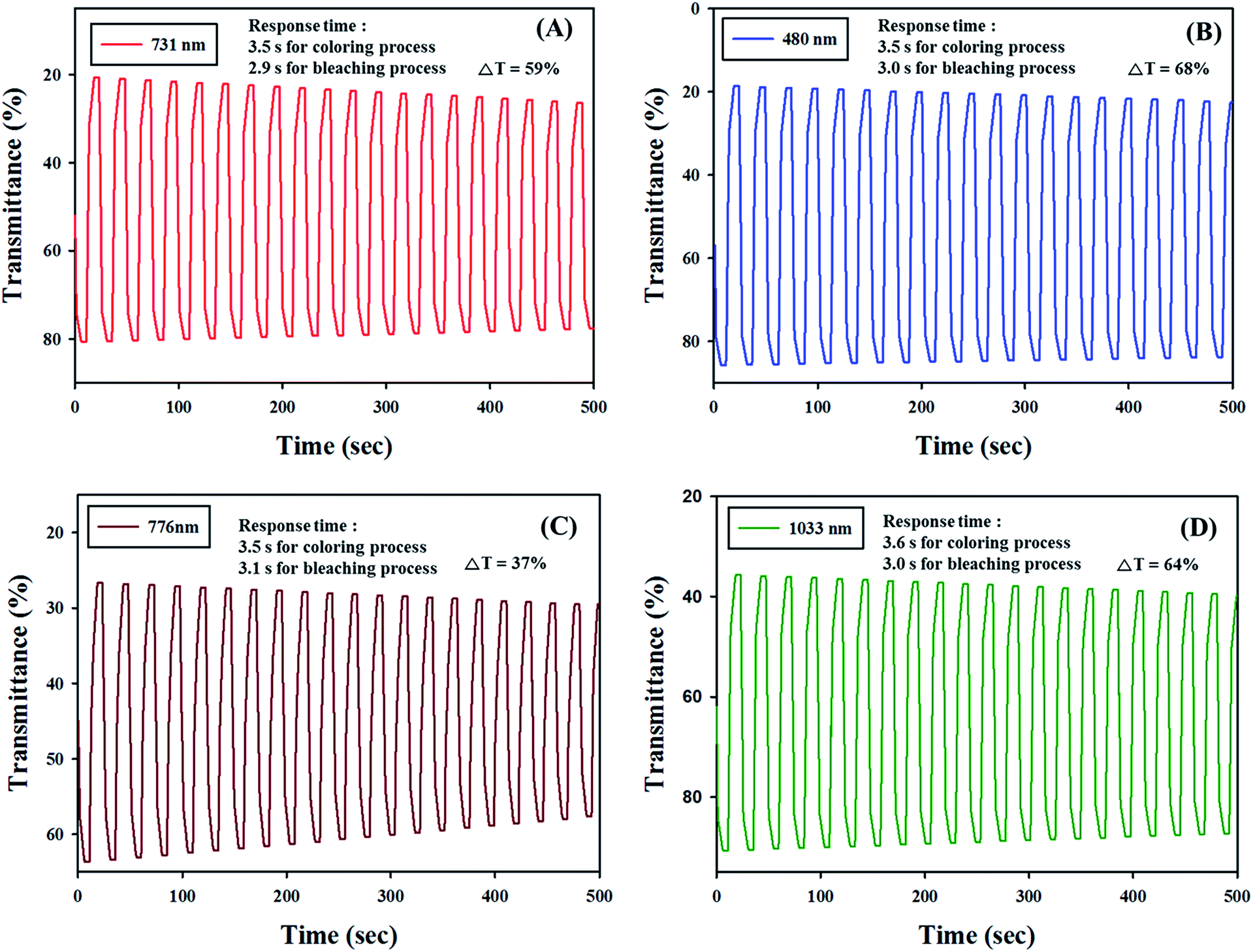
Electrochromic switching studies for the all polymers were done to monitor the % transmittance (T) as a function of time at their absorption maximum and to regulate the response time by stepping potential repeatedly between the neutral and oxidized states.43 The active area of the polymer film on ITO-glass is around 1 cm2 in TBABF4, in propylene carbonate. Fig. 6 represents the % T and current density changes of PJK1, PJK2, PJK3 and PJK4 as a function of the time at their longer wavelength absorption maximum, 731 nm for PJK1 with 0 to 2.5 V potential, 480 nm for PJK2, 776 nm for PJK3 and 1033 nm for PJK4 with 0 to 2.3 V potential by applying square-wave potential steps of 12 s (complete cycle time is 24 s) with 20 cycles. The optical contrast of the PJK1 was found to be 59% at 731 nm with a switching time of 3.5 seconds for coloring process and 2.9 seconds for bleaching process.44,45 PJK2, PJK3 and PJK4 exhibit electrochromic performance with contrast value (ΔT) 68%, 37%, 64% with switching time of 3.5 s/3 s, 3.5 s/3.1 s and 3.6 s/3.0 s respectively. From the data, it can be clearly found that PJK1, PJK2 and PJK4 were have the better stability and higher percent transmittance contrast than PJK3 after regular switching for 500 s (Fig. 6). Mainly PJK1 and PJK4 have the good optical contrasts in the NIR region, which is a very important property for various NIR applications, such as camouflage devices and polymer based field-effect transistors.
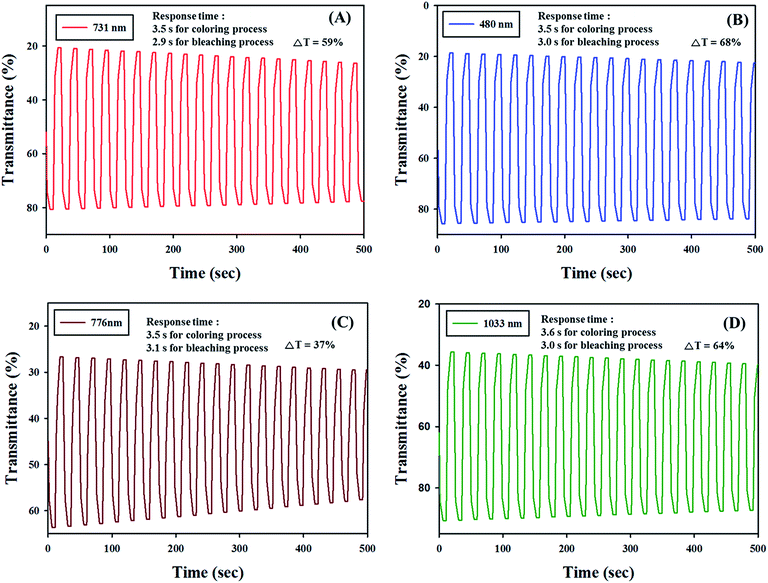 |
| | Fig. 6 Electrochromic switching under an applied square voltage signal for (A) PJK1 at 731 nm, (B) PJK2 at 480 nm, (C) PJK3 at 776 nm and (D) PJK4 at 1033 nm respectively, as a function of time. | |
3.7. Theoretical investigation
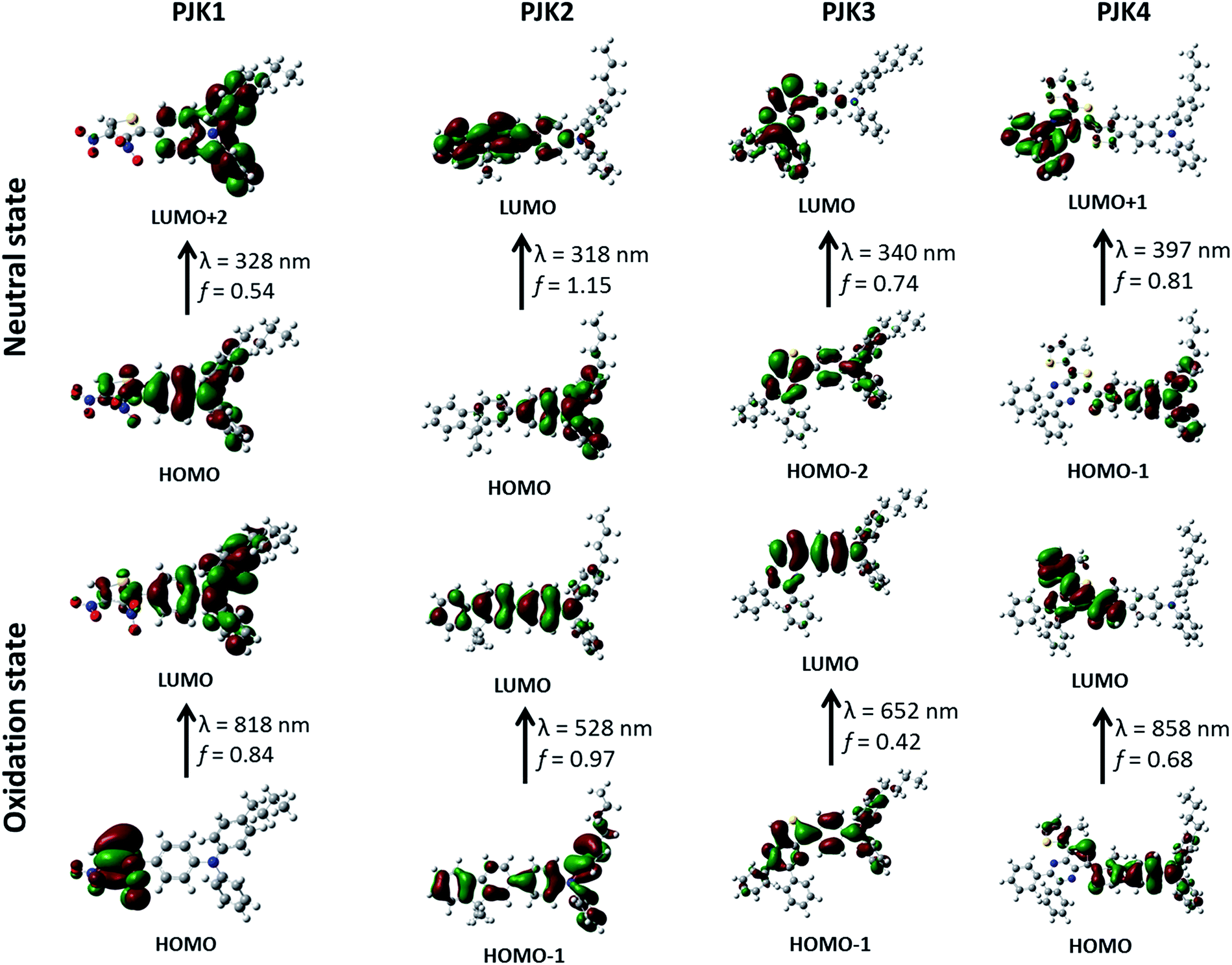
For this study, all the calculations were done for PJK polymer series using commercial package Gaussian 03.46 As shown in Fig. 7, the optimized geometries of monomers at neutral and oxidation states were obtained using Hartree–Fock (HF) combined with 3-21G* bases set.47 By using the semi-empirical quantum chemical method ZINDO including 25 lowest-energy electronic transitions, oscillator strength and absorption wavelength were generated.48
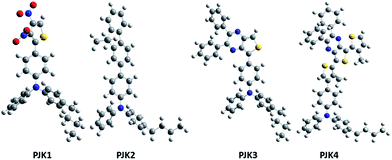 |
| | Fig. 7 Optimized geometries of monomers of conjugated polymers. | |
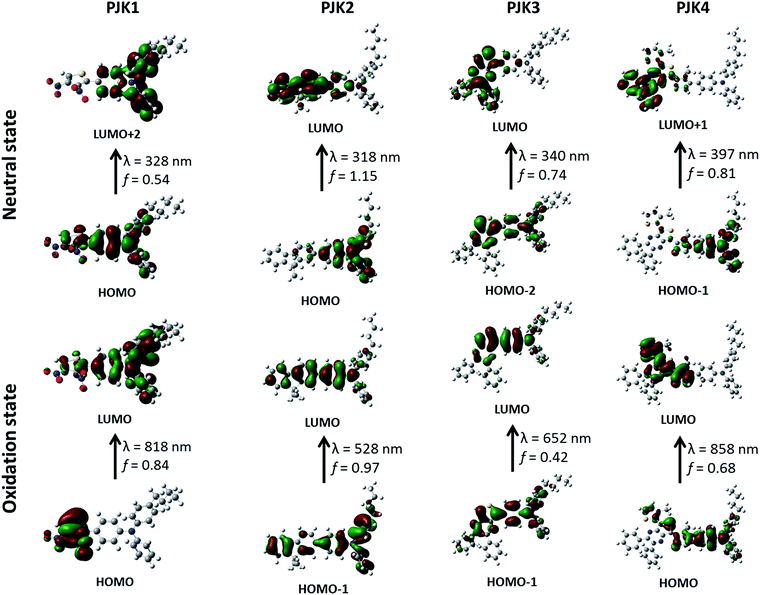
As shown in Table 2 and Fig. 8, optical (absorbance wavelength and oscillator strength) and transition properties in the neutral and oxidation states were used for investigation to better understand the optical transition of PJK materials. For the neutral state of PJK1, HOMO → LUMO+2 transition mainly dominates S0 → S7 transition with the absorption wavelength of 328 nm, which is contributed by charge transfer from the whole to the triarylamine part (Fig. 8). The transitions HOMO → LUMO, HOMO−2 → LUMO and HOMO−1 → LUMO+1 correspond to PJK2, PJK3 and PJK4 in the natural state mainly dominate S0 → S1, S0 → S3 and S0 → S5 electronic transitions having large oscillator strength with absorption at 318, 340 and 397 nm respectively. Which are contributed by charge transfer from triarylamine to the rest of the molecule. For the oxidation state of PJK1, PJK3 and PJK4, HOMO → LUMO, HOMO−1 → LUMO and HOMO → LUMO transitions respectively, mainly controls S0 → S1 transition with maximum absorption at 818, 652 and 858 nm, respectively. While, for PJK2 in the oxidation state, HOMO−1 → LUMO transition related to 528 nm mainly dominates S0 → S2 transition. The absorption wavelengths for the conjugated polymers are close to the experimental values (Fig. 5). Upon oxidation, the changes in the conjugation length in conjugated polymers PJK1, PJK2, PJK3 and PJK4 in the film state can be explained from this theoretical study. Thus, the NIR electrochromic phenomena of reported conjugated polymers can be successfully proved by theoretical investigation.
Table 2 The calculated absorption wavelength (λ), oscillator strength (f) and the corresponding MO transitions of monomers in neutral and oxidation states. The electronic transitions corresponding to the first excited state and the largest oscillator strength are shown
| |
Neutral state |
Oxidation state |
| State |
λ (nm) |
f (a.u.) |
Transition character |
State |
λ (nm) |
f (a.u.) |
Transition character |
| PJK1 |
S0 → S1 |
532 |
0.02 |
HOMO → LUMO+1 (11%) |
S0 → S1 |
818 |
0.84 |
HOMO → LUMO (91%) |
| S0 → S7 |
328 |
0.54 |
HOMO → LUMO+2 (49%) |
S0 → S2 |
614 |
0.37 |
HOMO−1 → LUMO (79%) |
| PJK2 |
S0 → S1 |
318 |
1.15 |
HOMO → LUMO (49%) |
S0 → S1 |
750 |
1.22 |
HOMO → LUMO (92%) |
| S0 → S19 |
222 |
1.02 |
HOMO−3 → LUMO (27%) |
S0 → S2 |
528 |
0.97 |
HOMO−1 → LUMO (89%) |
| PJK3 |
S0 → S1 |
482 |
0.36 |
HOMO → LUMO (78%) |
S0 → S1 |
652 |
0.42 |
HOMO−1 → LUMO (70%) |
| S0 → S3 |
340 |
0.74 |
HOMO−2 → LUMO (26%) |
S0 → S7 |
429 |
0.44 |
HOMO−5 → LUMO (41%) |
| PJK4 |
S0 → S1 |
544 |
0.44 |
HOMO → LUMO (42%) |
S0 → S1 |
858 |
0.68 |
HOMO → LUMO (65%) |
| S0 → S5 |
397 |
0.81 |
HOMO−1 → LUMO+1 (63%) |
S0 → S3 |
572 |
0.59 |
HOMO−2 → LUMO (37%) |
 |
| | Fig. 8 Electronic transitions of PJK1, PJK2, PJK3 and PJK4 in ground and excited states. | |
4. Conclusions
We have prepared four soluble, electrochromic triphenylamine-based copolymer derivatives and reported their fabrication into electrochromic cells using ITO-glass. The electrochromic devices were shown to exhibit good color changes when modulated between their neutral and oxidized states and color changes for PJK1, the color changed from orange to dark green; for PJK2, from light yellow to reddish brown; for PJK3, from light blue to grey; and for PJK4, from green to bluish green. These polymers revealed well switching times, is an important candidate for electrochromic display applications. These polymer electrochromic cells may be useful for conductive textiles, whose interesting features could find applications in adaptive camouflage.
Acknowledgements
This study was supported by the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT and Future Planning (Grant no. 2015063131).
Notes and references
- P. M. S. Monk, R. J. Mortimer and D. R. Rosseinsky, Electrochromism fundamentals and applications, VCH, Weinheim, Germany, 1995 Search PubMed.
- J. R. Platt, J. Chem. Phys., 1961, 34, 862–863 CrossRef CAS.
- R. G. Mortimer, Chem. Soc. Rev., 1997, 26, 147–156 RSC.
- H. J. Byker, Single-compartment, self-erasing, solution-phase electrochromic devices, solutions for use therein and uses thereof, US Pat., No. 4902108, Gentex Corp, 1990.
- D. S. K. Mudigonda, D. L. Meeker, D. C. Loveday, J. M. Osborn and J. P. Ferraris, Polymer, 1999, 40, 3407–3412 CrossRef CAS.
- P. M. S. Monk, J. Electroanal. Chem., 1997, 432, 175–179 CrossRef CAS.
- P. M. S. Monk, Handb. Lumin., Disp. Mater., Devices, 2003, 3, 261 CAS.
- W. C. D. Smith, Displays, 1982, 3, 67 CrossRef.
- R. J. Mortimer, Electrochim. Acta, 1999, 44, 2971–2981 CrossRef CAS.
- A. A. Argun, P.-H. Aubert, B. C. Thompson, I. Schwendeman, C. L. Gaupp, J. Hwang, N. J. Pinto, D. B. Tanner, A. G. M. Diarmid and J. R. Reynolds, Chem. Mater., 2004, 16, 4401–4412 CrossRef CAS.
- G. Sonmez, C. K. F. Shen, Y. Rubin and F. Wudl, Angew. Chem., Int. Ed., 2004, 43, 1498–1502 CrossRef CAS PubMed.
- H. Yu, S. Shao, L. Yan, H. Meng, Y. He, C. Yao, P. Xu, X. Zhang, W. Hu and W. Huang, J. Mater. Chem. C, 2016, 4, 2269–2273 RSC.
- D. Witker and J. R. Reynolds, Macromolecules, 2005, 38, 7636–7644 CrossRef CAS.
- D. M. Welsh, L. J. Kloeppner, L. Madrigal, M. R. Pinto, B. C. Thompson, K. S. Schanze, K. A. Abboud, D. Powell and J. R. Reynolds, Macromolecules, 2002, 35, 6517–6525 CrossRef CAS.
- W.-L. Yu, J. Pei, W. Huang and A. J. Heeger, Chem. Commun., 2000, 681–682 RSC.
- K. Ogino, A. Kanagae, R. Yamaguchi, H. Sato and J. Kurtaja, Macromol. Rapid Commun., 1999, 20, 103–106 CrossRef CAS.
- T.-H. Su, S.-H. Hsiao and G.-S. Liou, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 2085–2098 CrossRef CAS.
- M.-Y. Chou, M.-K. Leung, Y. O. Su, S. L. Chiang, C.-C. Lin, J.-H. Liu, C.-K. Kuo and C.-Y. Mou, Chem. Mater., 2001, 16, 654–661 CrossRef.
- S. H. Hsiao, G. S. Liou, Y. C. Kung and Y. M. Chang, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 2798–2809 CrossRef CAS.
- D. M. Welsh, L. J. Kloeppner, L. Madrigal, M. R. Pinto, B. C. Thompson, K. S. Schanze, K. A. Abbound, D. Powell and J. R. Reynolds, Macromolecules, 2002, 35, 6517–6525 CrossRef CAS.
- A. Cirpan, A. A. Argun, C. R. G. Grenier, B. D. Reeves and J. R. Reynolds, J. Mater. Chem., 2013, 13, 2422–2428 RSC.
- K. L. Wang, M. K. Leung, L. G. Hsieh, C. C. Chang, K. R. Lee, C. L. Wu, J. C. Jiang, C. Y. Tseng and H. T. Wang, Org. Electron., 2011, 12, 1048–1062 CrossRef CAS.
- X. H. Ouyang and Y. P. Huo, Appl. Phys. A, 2011, 105, 891–895 CrossRef CAS.
- S. Marco and J. Andres, J. Am. Chem. Soc., 2010, 132, 8372–8377 CrossRef PubMed.
- H. Hong, M. Y. Jo, Y. E. Ha and J. H. Kim, Macromol. Res., 2013, 21, 321–326 CrossRef CAS.
- Y.-L. Chu, C.-C. Cheng, Y.-C. Yen and F.-C. Chang, Adv. Mater., 2012, 24, 1894–1898 CrossRef CAS PubMed.
- R. M. Kellogag, A. P. Schaap, E. T. Harper and N. D. H. wynberg, J. Org. Chem., 1968, 33, 2902–2909 CrossRef.
- R. Mozingo, S. A. Harris, D. E. Wolf, C. E. Hoffhine, N. R. Easton and K. Folkers, J. Am. Chem. Soc., 1945, 67, 2092–2095 CrossRef CAS PubMed.
- H. Hailu, B. Atsbeha, S. Admassie, W. Mammo, V. J. T. Raju and Y. Chebude, Bull. Chem. Soc. Ethiop., 2011, 25, 221–231 CrossRef CAS.
- X. Liu, L. Li, J. Sun, Y. Yan, X. Shu, B. Liu, W. Sha, H. Feng, S. Sun and J. Zhu, Inorg. Chem., 2012, 51, 188–192 CrossRef CAS PubMed.
- M. Shahid, R. S. Ashraf, E. Klemm and S. Sensfuss, Macromolecules, 2006, 39, 7844–7853 CrossRef CAS.
- C. L. Chochos, S. P. Economopoulos, V. Deimede, V. G. Gregoriou, M. T. Lloyd, G. G. Malliaras and J. K. Kallitsis, J. Phys. Chem. C, 2007, 111, 10732–10740 CAS.
- C. V. Pharn, H. B. Mark and H. Zirnmer, Synth. Commun., 1986, 16, 689–696 CrossRef.
- R. Gong, Z. Guo, F. Li, Y. Song, Y. Mu, M. Li and X. Wan, Macromol. Chem. Phys., 2014, 215, 906–914 CrossRef CAS.
- M. He, T. M. Leslie and J. A. Sinicropi, Chem. Mater., 2002, 14, 4662–4668 CrossRef CAS.
- H. Usta, C. Risko, Z. Wang, H. Huang, M. K. Deliomeroglu, A. Zhukhovitskiy, A. Facchetti and T. J. Marks, J. Am. Chem. Soc., 2009, 131, 5586–5608 CrossRef CAS PubMed.
- J. Bras, S. Guillerez and B. P. Donat, Chem. Mater., 2000, 12, 2372–2384 CrossRef CAS.
- S. Beaupré, A.-C. Breton, J. Dumas and M. Leclerc, Chem. Mater., 2009, 21, 1504–1513 CrossRef.
- S. Beaupré, J. Dumas and M. Leclerc, Chem. Mater., 2006, 18, 4011–4018 CrossRef.
- B. D. Reeves, C. R. G. Grenier, A. A. Argun, A. Cirpan, T. D. McCarley and J. R. Reynolds, Macromolecules, 2004, 37, 7559–7569 CrossRef CAS.
- G. Sonmez, H. B. Sonmez, C. K. F. Shen, R. W. Jost, Y. Rubin and F. Wudl, Macromolecules, 2005, 38, 669–675 CrossRef CAS.
- C. Fan, C. Ye, X. Wang, Z. Chen, Y. Zhou, Z. Liang and X. Tao, Macromolecules, 2015, 48, 6465–6473 CrossRef CAS.
- M. Yin, Y. Yan, F. Li, X. Liu, C. Wang and D. Chao, RSC Adv., 2016, 6, 50529–50533 RSC.
- G. Atakana and G. Gunbas, RSC Adv., 2016, 6, 25620–25623 RSC.
- Z. P. Xu, X. M. Chen, S. Mi, J. M. Zheng and C. Y. Xu, Org. Electron., 2015, 26, 129–136 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery Jr, T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 03, Revision B.04, Gaussian Inc., Pittsburgh, PA, 2003 Search PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- S. Nachimuthu, K.-H. Lai, F. Taufany and J.-C. Jiang, Phys. Chem. Chem. Phys., 2014, 16, 15389–15399 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed 1H NMR, 13C NMR, HRMS and IR data available. See DOI: 10.1039/c6ra12112h |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.