
Computational study of the binding orientation and affinity of PPARγ agonists: inclusion of ligand-induced fit by cross-docking†
Camila Muñoz-Gutierreza,
Francisco Adasme-Carreñoa,
Eduardo Fuentesb,
Iván Palomob and
Julio Caballero*a
aCentro de Bioinformatica y Simulacion Molecular (CBSM), Universidad de Talca, 2 Norte 685, Talca, Chile. E-mail: jcaballero@utalca.cl
bDepartment of Clinical Biochemistry and Immunohematology, Faculty of Health Sciences, Interdisciplinary Excellence Research Program on Healthy Aging (PIEI-ES), Talca University, 2 Norte 685, Talca, Chile
First published on 30th June 2016
Abstract
The peroxisome proliferator-activated receptors (PPARs) comprise a family of three nuclear receptor isoforms (γ, β/δ, α) which are key regulators of metabolism and inflammation. A series of potent PPARγ agonists with high chemical diversity and variable activities have been reported in recent years; however, few molecular atomistic studies have been carried out to describe the formation of the complexes. In this work, the docking of several potent agonists (organized into three sets) were performed inside the binding site of PPARγ and quantitative correlations between the obtained scoring energy functions and experimental biological activities were determined using the Glide and MM/GBSA methods. In silico experiments were achieved using a cross-docking protocol which includes sixteen PPARγ crystallographic structures. The studied ligands were positioned at the previously described binding pocket establishing interchangeable hydrogen bonds with key residues. Significant correlations (R2 > 0.6) were reported for the three studied sets using different methods. The use of several representative protein conformations for cross-docking, indicates that the induced-fit effects on the residues in the binding site have to be considered to plan docking experiments in PPARγ.
Introduction
The World Health Organization estimated that 30% of all deaths worldwide are due to cardiovascular diseases (CVDs).1 In most cases, CVDs are the consequence of an atherosclerotic plaque rupture and thrombus formation. The role of platelets in thrombus formation is well understood and is a primary factor in acute coronary syndromes.2,3 After atheromatous plaque rupture, platelets adhere, secrete their granule contents, aggregate, and initiate thrombus formation.4,5 Furthermore, platelets have been determined to have an expanded function in atherosclerosis, triggering early events.6 Peroxisome proliferator-activated receptors (PPARs), such as PPARγ, β/δ, and α,7 are nuclear receptor proteins widely expressed in the cardiovascular system. They are key metabolism and inflammation regulators; therefore, they play an important role in the processes that govern chronic inflammatory diseases.8,9 Several reports refer to the PPARs regulatory role in atherogenesis.10,11 PPARs are expressed in human platelets, which are involved in inflammation processes, thus contributing to haemostasis, where abnormal platelet adhesion/activation can lead to formation of a clot, namely thrombosis.12,13Several studies have demonstrated that drugs that affect PPARs activation may exert antiplatelet activities, and this would, at least partly, explain the inhibitory effects of statins (secondary PPARs activators), such as simvastatin, on platelets activation.14,15 Particularly, it has been suggested that PPARγ agonists reduce the development of atherosclerosis and myocardial ischemia–reperfusion injury; both of these conditions are associated with abnormal platelet activation.16 Among the multiple signaling molecules regulating platelet activation, cAMP plays a crucial role in the pathway, where its intracellular levels inhibit platelet aggregation.17,18 It has been reported that PPAR activation by ligand binding increases cAMP concentration due to PKC repression, which allows greater activity of adenylyl cyclase (ATP to cAMP).19
A large number of PPARγ pharmacomodulators have been synthesized, which share basically a two-module structural scaffold: an activation head group that forms hydrogen bonds (HBs) with key residues and a hydrophobic tail group. Despite the high structural variability of PPARγ ligands, it has been reported that most PPARγ agonists establish, at least, a HB network with the polar aromatic residues His323, Tyr327, His449, and Tyr473.18–20 The largely hydrophilic zone formed by these residues is important for the binding of all strong agonists and accommodates polar head groups such as carboxylate and thiazolidinedione (TZD), common on PPARγ modulators.18
Due to the lack of theoretical studies on PPARγ ligand binding, we have set out to construct computational models which correlate the calculated binding energies of three series of potent chemically diverse PPARγ agonists with their experimental biological activities21–23 through a protocol combining cross-docking and re-scoring calculations. This protocol provides great statistical correlations and insights about PPARγ agonists binding at atomistic level.
Materials and methods
Dataset
Three series of chemically-diverse PPARγ full agonists reported in the literature (Fig. 1, Tables 1–3) were studied by means of computational chemistry methods in order to elucidate the molecular basis for their binding behavior. Set 1 comprised 60 substituted indanylacetic acids (compounds A1–A60 in Fig. 1 and Table 1), which showed EC50 biological activities at the nanomolar range measured via Fluorescence Resonance Energy Transfer (FRET) assays.21 Set 2 has 40 anthranilic acid derivatives with EC50 measured in PPARγ transactivation assays (compounds B1–B40 in Fig. 1 and Table 2).22 Finally, Set 3 contains 50 antidiabetic agents and comprises TZD analogues: 5-[4-(2-phenylethoxy)benzyl]-1,3-thiazolidine-2,4-dione derivatives, 5-benzyl-1,3-thiazolidine-2,4-dione derivatives, and 5-(pyridine-3-ylmethyl)-1,3-thiazolidine-2,4-dione derivatives (compounds C1–C50 in Fig. 1 and Table 3).23 For this ligand set, antihyperglycemic activities (lnHMGA) were reported, which were obtained using genetically obese and diabetic yellows KK mice experiments. In a more recent work, Kumar et al. proposed that the antidiabetic activities of these compounds is due to PPARγ full agonistic activities.24 This proposal is sound, since TZD analogues have been identified as specific PPARγ agonists in previous literature.25,26 Considering the foregoing, our modeling assumes that antihyperglycemic activities are correlated with PPARγ agonistic potencies and other targets do not interfere with the relative potencies of the compound activities.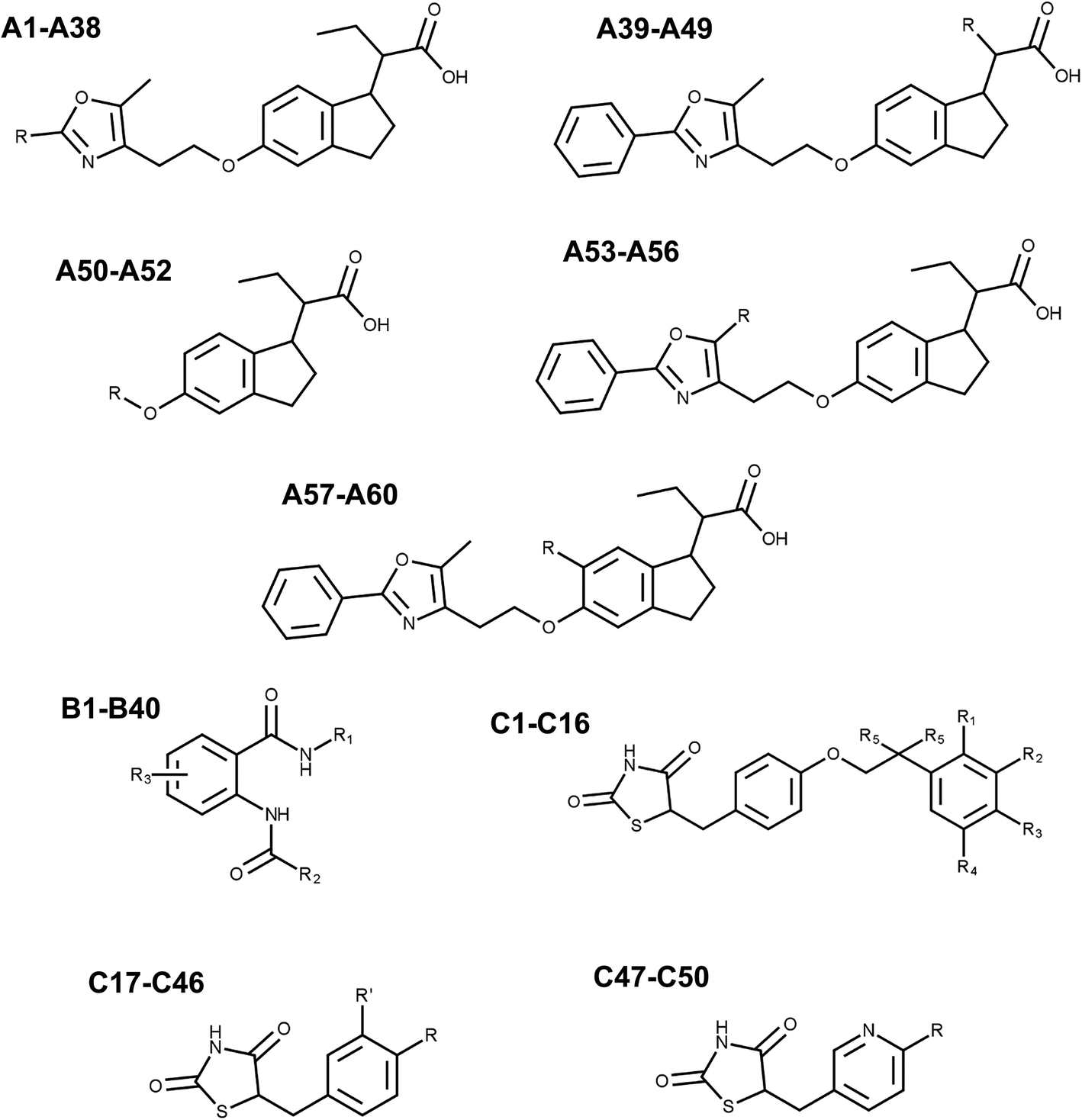 | ||
| Fig. 1 PPARγ agonist sets. General structures of compounds of the Set 1 (A1–A60), Set 2 (B1–B40) and Set 3 (C1–C50). | ||
| Compound | R | EC50 [nM] |
|---|---|---|
| A1 | Benzyl | 330 |
| A2 | (4-Fluoro)benzyl | 523 |
| A3 | Phenoxymethyl | 45 |
| A4 | (4-Chlorophenoxy)methyl | 104 |
| A5 | Cyclopentyl | 212 |
| A6 | Cyclohexyl | 136 |
| A7 | Phenyl | 42 |
| A8 | 2-Methylphenyl | 153 |
| A9 | 3-Methylphenyl | 34 |
| A10 | 4-Methylphenyl | 45 |
| A11 | 4-Ethylphenyl | 40 |
| A12 | 4-Isopropylphenyl | 22 |
| A13 | 4-(n-Butyl)phenyl | 16 |
| A14 | 4-(tert-Butyl)phenyl | 19 |
| A15 | 3-Methoxyphenyl | 1 |
| A16 | 4-Methoxyphenyl | 42 |
| A17 | 2-Fluorophenyl | 49 |
| A18 | 3-Fluorophenyl | 61 |
| A19 | 4-Fluorophenyl | 59 |
| A20 | 4-Chlorophenyl | 206 |
| A21 | 3-Trifluoromethyl | 21 |
| A22 | 4-Trifluoromethyl | 205 |
| A23 | 3,4-Dimethylphenyl | 32 |
| A24 | 3,5-Bis(trifluoromethyl)phenyl | 247 |
| A25 | 3-Fluoro-4-methylphenyl | 43 |
| A26 | 4-Fluoro-3-methylphenyl | 77 |
| A27 | 3,4-Dimethoxy | 84 |
| A28 | 3,4-(Methylenedioxy)phenyl | 14 |
| A29 | 2,6-Difluorophenyl | 368 |
| A30 | 2.4-Dichlorophenyl | 738 |
| A31 | 3.4-Dichlorophenyl | 94 |
| A32 | (4-Phenyl)phenyl | 43 |
| A33 | 1-Naphthyl | 113 |
| A34 | 2-Naphthyl | 16 |
| A35 | 2-Furyl | 396 |
| A36 | 6-(Dihydrobenzofuranyl) | 6 |
| A37 | 2-Benzothienyl | 37 |
| A38 | 3-(5-Methyl)isoxazoyl | 762 |
| A39 | Methyl | 104 |
| A40 | gem-Dimethyl | 5640 |
| A41 | 2,2,2-Trifluoroethyl | 177 |
| A42 | Cyclopropyl | 160 |
| A43 | Propyl | 52 |
| A44 | 3-Phenylpropyl | 3180 |
| A45 | Phenyl | 9560 |
| A46 | Carboxy | 1860 |
| A47 | 2-Carboxyethyl | 68 |
| A48 | Methoxy | 45 |
| A49 | Ethoxy | 12 |
| A50 | 2-(5-Methyl-2-phenylthiazol-4-yl)ethyl | 85 |
| A51 | 2-(5-Methyl-3-phenyl-1H-pyrazol-1-yl)ethyl | 196 |
| A52 | (5-Methyl-2-phenyloxazol-4-yl)methyl | 632 |
| A53 | Hydrogen | 306 |
| A54 | Ethyl | 46 |
| A55 | Isopropyl | 347 |
| A56 | Phenyl | 617 |
| A57 | Chloro | 294 |
| A58 | Phenyl | 290 |
| A59 | 4-Chlorophenyl | 168 |
| A60 | 4-Methoxyphenyl | 124 |
| Compounds | R1 | R2 | R3 | EC50 [μM] |
|---|---|---|---|---|
| B1 | 3-Carboxypropyl | 2-Naphthyl | — | 18.1 |
| B2 | 3-Carboxypropyl | 1-Naphthyl | — | 41.0 |
| B3 | 3-Carboxypropyl | p-Ethylphenyl | — | 39.0 |
| B4 | 3-Carboxypropyl | 4-Biphenylyl | — | 13.0 |
| B5 | 3-Carboxypropyl | 3-Biphenylyl | — | 37.0 |
| B6 | 3-Carboxypropyl | 2-Biphenylyl | — | 23.0 |
| B7 | 4-Carboxybutyl | 2-Naphthyl | — | 26.0 |
| B8 | 5-Carboxypentyl | 2-Naphthyl | — | 40.0 |
| B9 | m-Carboxyphenyl | 2-Naphthyl | — | 5.5 |
| B10 | p-Carboxyphenyl | 2-Naphthyl | — | 21.0 |
| B11 | p-(Carboxymethyl)phenyl | 2-Naphthyl | — | 25.0 |
| B12 | (p-Carboxyphenyl)methyl | 2-Naphthyl | — | 56.0 |
| B13 | m-Carboxyphenyl | 1,3-Dioxa-5-indanyl | — | 25.0 |
| B14 | m-Carboxyphenyl | 3-Fluoro-4-(trifluoromethyl)phenyl | — | 6.9 |
| B15 | m-Carboxyphenyl | 3,5-Bis(trifluoromethyl)phenyl | — | 11.0 |
| B16 | m-Carboxyphenyl | p-(tert-Butyl)phenyl | — | 3.9 |
| B17 | m-Carboxyphenyl | p-(Trifluoromethyl)phenyl | — | 8.5 |
| B18 | m-Carboxyphenyl | p-Bromophenyl | — | 4.5 |
| B19 | m-Carboxyphenyl | p-(tert-Butyl)phenyl | 5-Cl | 5.4 |
| B20 | m-Carboxyphenyl | p-(tert-Butyl)phenyl | 4-Cl | 9.0 |
| B21 | m-(Carboxymethyl)phenyl | p-(tert-Butyl)phenyl | — | 6.2 |
| B22 | m-(2-Carboxyethyl)phenyl | p-(tert-Butyl)phenyl | — | 5.0 |
| B23 | 3-Carboxy-2-methylphenyl | p-(tert-Butyl)phenyl | — | 13.5 |
| B24 | 3-Carboxy-4-methylphenyl | p-(tert-Butyl)phenyl | — | 5.0 |
| B25 | 5-Carboxy-2-methylphenyl | p-(tert-Butyl)phenyl | — | 6.3 |
| B26 | 3-Carboxy-4-methoxyphenyl | p-(tert-Butyl)phenyl | — | 4.4 |
| B27 | 3-Carboxy-4-chlorophenyl | p-(tert-Butyl)phenyl | — | 5.4 |
| B28 | 4-Bromo-3-carboxyphenyl | p-(tert-Butyl)phenyl | — | 6.3 |
| B29 | 5-Carboxy-2-methoxyphenyl | p-(tert-Butyl)phenyl | — | 4.5 |
| B30 | 5-Carboxy-2-chlorophenyl | p-(tert-Butyl)phenyl | — | 1.9 |
| B31 | 5-Carboxy-2-fluorophenyl | p-(tert-Butyl)phenyl | — | 5.8 |
| B32 | m-Carboxyphenyl | p-(Trifluoromethyl)phenyl | 5-Cl | 4.7 |
| B33 | m-Carboxyphenyl | p-(Trifluoromethyl)phenyl | 4-Cl | 8.7 |
| B34 | m-(Carboxymethyl)phenyl | p-(Trifluoromethyl)phenyl | — | 9.4 |
| B35 | m-(2-Carboxyethyl)phenyl | p-(Trifluoromethyl)phenyl | — | 6.9 |
| B36 | m-(Carboxymethyl)phenyl | 4-Biphenylyl | — | 2.6 |
| B37 | m-(Carboxymethyl)phenyl | p-(Methylthio)phenyl | — | 17.4 |
| B38 | m-(Carboxymethyl)phenyl | p-(Trifluoromethylthio)phenyl | — | 4.4 |
| B39 | m-(Carboxymethyl)phenyl | p-Methoxyphenyl | — | 13.2 |
| B40 | m-(Carboxymethyl)phenyl | 2-Naphthyl | — | 18.0 |
| C1–C16 | R1 | R2 | R3 | R4 | R5 | lnMHGA [μM] |
|---|---|---|---|---|---|---|
| C1 | H | H | H | H | Me | −4.773 |
| C2 | Methoxy | H | H | H | Me | −4.855 |
| C3 | H | H | Ethyl | H | Me | −4.849 |
| C4 | H | H | Ethoxy | H | Me | −4.890 |
| C5 | H | H | H | H | H | −4.691 |
| C6 | H | H | Me | H | H | −4.733 |
| C7 | Methoxy | H | H | H | H | −4.779 |
| C8 | H | H | Ethyl | H | H | −4.773 |
| C9 | H | H | Ethoxy | H | H | −4.818 |
| C10 | H | H | Cl | H | H | −4.790 |
| C11 | Methoxy | H | Methyl | H | H | −4.818 |
| C12 | H | Methoxy | Methoxy | Methoxy | H | −4.934 |
| C13 | H | 1,3-Dioxolane | H | H | −4.818 | |
| C14 | H | Methoxy | H | H | Me | −4.854 |
| C15 | H | H | Methoxy | H | H | −4.779 |
| C16 | H | Methoxy | Methoxy | H | H | −4.860 |
| C17–C46 | R | R′ | lnMHGA [μM] |
|---|---|---|---|
| C17 | 3-Phenylpropoxy | — | −5.139 |
| C18 | 4-Phenylbutoxy | — | −5.179 |
| C19 | 2-Phenylpropoxy | — | −4.733 |
| C20 | 1-Methyl-2-phenylethoxy | — | −4.733 |
| C21 | 2-Methyl-2-phenylpropoxy | — | −4.812 |
| C22 | 2,2-Dimethylhexyloxy | — | −4.715 |
| C23 | 2,2-Dimethylheptyl | — | −5.162 |
| C24 | m-Chlorophenoxy | — | −5.115 |
| C25 | Neopentyloxy | — | −4.987 |
| C26 | (1-Methylcyclohexyl)methoxy | — | −4.700 |
| C27 | (1-Propylcyclohexyl)methoxy | — | −4.790 |
| C28 | (3-Pyridyl)methoxy | — | −4.651 |
| C29 | Phenethoxy | — | −4.694 |
| C30 | 3-(3-Pyridyl)propoxy | — | −4.736 |
| C31 | 2-(2-Thienyl)ethoxy | — | −4.709 |
| C32 | 2-(2-Furyl)ethoxy | — | −5.066 |
| C33 | 2-(Isopropylamino)ethoxy | — | −5.730 |
| C34 | 2-(1-Propylbutylamino)ethoxy | — | −5.165 |
| C35 | 2-(1-Pyrrolidinyl)ethoxy | — | −4.670 |
| C36 | 2-Piperidinoethoxy | — | −4.712 |
| C37 | Amino | — | −5.236 |
| C38 | Dioxy-p-tolylthioamino | — | −4.709 |
| C39 | Methoxy | Methoxy | −5.587 |
| C40 | 2,2-Diphenylpropoxy | — | −5.341 |
| C41 | 2,2-Dimethylpentyloxy | — | −5.078 |
| C42 | (1-Methylcyclopentyl)methoxy | — | −4.668 |
| C43 | p-Chlorophenoxy | — | −5.156 |
| C44 | 2-(6-Methyl-2-pyridyl)ethoxy | — | −4.737 |
| C45 | 2-(tert-Butylamino)ethoxy | — | −5.776 |
| C46 | Propionylamino | — | −4.935 |
| C47–C50 | R | lnMHGA [μM] |
|---|---|---|
| C47 | 2-Cyclohexylethoxy | −5.118 |
| C48 | 2-Morpholinoethoxy | −5.820 |
| C49 | Phenethoxy | −4.697 |
| C50 | (1-Methylcyclohexyl)methoxy | −4.712 |
Protein and ligand preparations
The tridimensional (3D) PPARγ X-ray crystallographic structures in Protein Data Bank (PDB) were inspected, and those co-crystallized with full agonists and whose structure is complete (e.g., no missing loops) were selected. Sixteen PPARγ co-crystallized with different ligands were obtained; PDB codes are 2ATH (resolution: 2.28 Å), 2F4B (resolution: 2.07 Å), 2I4J (resolution: 2.10 Å), 2PRG (resolution: 2.30 Å), 2Q59 (resolution: 2.20 Å), 2VV0 (resolution: 2.55 Å), 2VV1 (resolution: 2.20 Å), 2VV2 (resolution: 2.75 Å), 2XKW (resolution: 2.02 Å), 3B3K (resolution: 2.60 Å), 3CDS (resolution: 2.65 Å), 3GBK (resolution: 2.30 Å), 3HO0 (resolution: 2.60 Å), 3HOD (resolution: 2.10 Å), 3NOA (resolution: 1.98 Å), and 3QT0 (resolution: 2.50 Å). The Protein Preparation Wizard module included in the Schrödinger Suite 2015-1 was employed for preparing the protein structures, where all X-ray waters were removed, regardless of the complex, since no water molecules were conserved in the binding sites. The HB network was optimized at neutral pH by sampling rotamers, protonation states and tautomers for Asn, Asp, Gln, Glu and His residues, including hydroxyls and thiols terminal groups as well. Structures were then relaxed by means of a restrained molecular minimization using the Impact Refinement (Impref) module,27 with the heavy atoms restrained to remain within a root-mean-square deviation (RMSD) of 0.30 Å from the initial coordinates. This step allows hydrogen atoms to be freely minimized, while allowing for heavy-atom movement to relax strained bonds, angles, and steric clashes.3D ligand molecular structures were sketched using the chemical editor included in Maestro software (Maestro, version 10.1, Schrödinger, LLC, New York, NY, 2015). Then, they were prepared using the LigPrep module (LigPrep, version 3.3, Schrödinger, LLC, New York, NY, 2015), where the ionization/tautomeric states were predicted at physiological pH conditions using Epik (Epik, version 3.1, Schrödinger, LLC, New York, NY, 2015). Finally, they were energy minimized using Macromodel (MacroModel, version 10.7, Schrödinger, LLC, New York, NY, 2015) with the OPLS2005 force field to obtain relaxed ligand structures.
Docking calculations
Molecular docking calculations were performed using the Glide program (MacroModel, version 10.7, Schrödinger, LLC, New York, NY, 2015), employing Standard Precision (SP) and Extra Precision (XP) algorithms.28 Glide uses a series of hierarchical filters to search for possible ligand binding locations in a selected cavity of the receptor. The hierarchy begins with a site-point search. After this, diameter and subset tests are performed (selection of possible orientations of the ligand diameter and rotations about the ligand diameter). Then, atoms capable of making HBs are scored; if this score is good all interactions with the receptor are also scored with a discretized version of the ChemScore.29 Poses obtained are refined and minimized on the van der Waals and electrostatic grids of the receptor. Finally, minimized poses are re-scored using GlideScore scoring function, which is based on ChemScore, but includes steric-clash and buried polar terms to penalize electrostatic mismatches.30 SP mode includes the GlideScore scoring function as described above, but XP mode has a more sophisticated scoring function (GlideScore XP), which includes additional terms over the SP scoring function and a more complete treatment of some of the SP terms.28 The key characteristics of GlideScore XP scoring are (i) the application of large desolvation penalties to both protein and ligand polar and charged groups in a variety of suitable cases, and (ii) the identification of specific chemical features that provide very large contributions to enhanced binding affinity.XP mode provides a better sampling protocol and additional/improved scoring terms such as the hydrophobic reward term, better treatment of HBs, and enhanced detection of π-stacking interactions. However, XP docking mode is much stricter than SP in order to rule out false positives, but it often discards active conformations if they are not compatible with the particular conformation of the receptor that is being used, effectively outputting fewer binding poses. Considering the large chemical diversity of the chosen ligand sets, a cross-docking strategy was employed to account for receptor flexibility upon ligand binding by carrying out the docking calculations on the sixteen crystal structures mentioned above. Docking grids were generated with the default settings using the ligand in the binding site as centroid while ensuring that the grid box size was big enough to cover the entire receptor binding site. Default docking parameters were used, and no constraints were included. All the 150 compounds were docked inside the sixteen PPARγ crystal structures; at most ten docking ligand poses were retained per run. The best pose for each run was selected as described below.
A crucial point in the development of good practices in docking for evaluation of ligand orientations is the previous definition of the Essential Chemical Interactions Described for Analogue Ligands (ECIDALs) found in the reported crystallographic data in PDB. In common docking protocols focused to identify ligand orientations, the binding pose showing the most negative binding energy is selected according to the maximum score; however, in most of docking experiments, the binding poses showing the most negative energies frequently do not comply with ECIDALs. It is more reasonable to select the pose with the most negative energy which complies with ECIDALs. The vast majority of the known PPARγ agonists establish HB interactions with the tetrad of aromatic residues (His323 and Tyr327 on helix H5, His449 on helix H11, and Tyr473 on helix H12);18–20 therefore, this feature was defined as the ECIDALs in our study. After selecting one pose for each case, 2048 ligand–protein complexes were obtained, but cases which did not yield poses with ECIDALs had no representation.
Pose re-scoring via MM/GBSA
MM/GBSA free energy calculations were applied using the method Prime MM/GBSA (Prime, version 3.9, Schrödinger, LLC, New York, NY, 2015) to the complexes that have ECIDALs representation in docking experiments. Prime MM/GBSA is an efficient method to rapidly refine and rescore docking results which combines molecular mechanics energy and implicit solvation models at a reasonable computational cost. This method estimates the binding energy (ΔEbinding) as the difference between the energy of the bound complex and the energy of the unbound protein and ligand. It considers binding energy decomposition into contributions originating from different physico-chemical energy terms. Specifically, these terms are calculated for the protein–ligand complex, for the ligand, and for the protein. ΔEbinding is computed using to the following equation:| ΔEbinding = ΔEMM + ΔEsolv | (1) |
During Prime MM/GBSA calculations, the variable dielectric solvent model VSGB 2.0 (ref. 31) was employed, which includes empirical corrections to model directionality of HB interactions and π stacking interactions. Residues within 5.0 Å from the ligand were allowed to be flexible during the minimization of the complexes, keeping the rest of the structure fixed, to relieve minor steric clashes between the docked ligand and nearby side chains. Prime MM/GBSA demonstrated excellent results for the binding free energies estimations of a wide range of protein–ligand complexes.32–36
Correlation analysis
After docking and MM/GBSA re-scoring applications, several poses for each ligand were obtained. Each pose contains a ligand–protein pair, where the ligand is one of the compounds under study and the protein is one of the sixteen PPARγ 3D X-ray crystallographic structures. An in-house Python script (described in the ESI, Section S1 and Fig. S1†) was applied to select a representative complex for each ligand that better adjust global correlations between calculated energy values (obtained from Glide scoring or MM/GBSA) and logarithmic biological activities of compounds from the sets 1, 2 and 3. As a result, this algorithm gives the ligand–protein pairs which produce the highest correlations. A scheme of the complete protocol is shown in Fig. 2. | ||
| Fig. 2 Cross-docking workflow diagram. | ||
Results and discussion
Selection of the successful models
The protocol described in Materials and methods section yielded two poses for each compound using Glide SP and XP, and two additional poses after re-scoring SP and XP results using MM/GBSA. These poses were selected by optimizing the correlations between calculated scoring energies and experimental logarithmic activities for the three studied sets. In this sense, combination of the calculated energy values for the selected poses gives the higher correlation R2 value, where successful results were achieved when R2 > 0.5.For all sets, rather good correlations with the biological activities were obtained (Table 4), indicating a success in explaining the experimental structure–activity relationships through the proposed protocol. Docking poses obtained in different crystallographic structures were chosen to construct the best correlations. Table 5 shows the complete list of the selected crystal-‘docked pose’ combinations for each set using the four different approaches. The correlation coefficients obtained from the Glide scoring function in the SP docking mode were greater than 0.6 in all cases, but the usage of more accurate approaches such as the XP docking algorithm or the MM/GBSA method did not always end up in a better ranking performance.
| Set | N | Activity | N° crystals | R2 for Glide scoringb | R2 for Prime MM/GBSAb | |||
|---|---|---|---|---|---|---|---|---|
| Type | Range | SP | XP | SP | XP | |||
| a N indicates the number of compounds in the data set, N° crystals corresponds to the number of receptor crystal conformations used in the correlation models.b The best structure–activity relationship models are marked in bold. | ||||||||
| 1 | 60 | EC50 | 1 to 9560 nM | 4–9 | 0.63 | 0.57 | 0.56 | 0.66 |
| 2 | 40 | EC50 | 1.9 to 56 μM | 8–10 | 0.66 | 0.59 | 0.76 | 0.79 |
| 3 | 50 | lnHMGA | −5.82 to −4.65 | 8–10 | 0.64 | 0.52 | 0.46 | 0.49 |
| Model | Set 1 | Set 2 | Set 3 | |||
|---|---|---|---|---|---|---|
| PDB | Molecules | PDB | Molecules | PDB | Molecules | |
| Glide SP | 2Q59 | A3 A7 A9 A10 A11 A12 A13 A14 A15 A16 A17 A18 A19 A21 A23 A25 A28 A34 A36 A37 A43 A48 A54 | 2ATH | B6 B7 B8 B13 B23 | 2ATH | C4 C9 C11 C13 C17 C32 |
| 2VV0 | A1 A2 A5 A8 A20 A22 A24 A29 A30 A33 A35 A38 A41 A42 A44 A45 A46 A51 A53 | 2F4B | B2 B4 B15 B24 B25 B27 B28 | 2PRG | C1 C6 C8 C10 C15 C19 C20 C21 C22 C23 C25 C26 C30 C36 C37 C42 C46 | |
| 2VV2 | A6 A32 A40 A49 A50 A52 A57 | 2I4J | B19 B29 | 2Q59 | C2 C7 C27 C38 C50 | |
| 3NOA | A4 A26 A27 A31 A39 A47 A55 A56 A58 A59 A60 | 2PRG | B16 B20 B32 | 2VV0 | C33 C34 C45 | |
| 2VV1 | B21 B22 B26 B30 B31 B33 B35 B36 | 2VV1 | C39 | |||
| 2XKW | B1 B12 B39 B40 | 3B3K | C3 C5 C14 C28 C29 C31 C35 C40 C44 C49 | |||
| 3CDS | B5 B11 | 3CDS | C12 C41 C48 | |||
| 3GBK | B9 B14 B17 B18 B34 B38 | 3QT0 | C16 C18 C24 C43 C47 | |||
| 3HO0 | B3 | |||||
| 3HOD | B10 B37 | |||||
| Glide XP | 2I4J | A6 A7 A10 A18 A29 A31 A36 A41 A57 | 2F4B | B5 B15 B26 B37 | 2F4B | C4 C16 C37 C40 C43 |
| 2PRG | A38 A40 A53 | 2PRG | B12 B20 | 2PRG | C7 C10 C14 C23 C25 C27 C46 | |
| 2Q59 | A11 A16 A19 A25 A28 A34 A39 A43 A45 A47 A49 A50 A54 A60 | 2Q59 | B7 B10 B23 B25 B30 B32 B35 B38 | 2VV1 | C24 C39 | |
| 2VV0 | A3 A4 A8 A20 A30 A33 A42 A55 | 2VV1 | B16 B17 B21 B22 B29 B31 B33 B34 | 2VV2 | C1 C2 C5 C8 C9 C11 C13 C15 C19 C20 C22 C26 C28 C29 C30 C31 C36 C38 C42 C47 C50 | |
| 2VV1 | A27 A35 A44 A58 | 2XKW | B1 B2 B3 B6 B11 B40 | 2XKW | C3 C6 C21 C35 C44 C45 C49 | |
| 2VV2 | A2 A5 A22 A24 A46 A51 | 3GBK | B9 B13 B14 B18 B19 B27 B28 B36 B39 | 3CDS | C33 | |
| 3GBK | A1 A9 A12 A13 A14 A15 A17 A21 A23 A26 A32 A37 A48 A59 | 3HO0 | B8 | 3HOD | C18 C34 C41 C48 | |
| 3HO0 | A56 | 3NOA | B4 B24 | 3NOA | C12 C17 C32 | |
| 3HOD | A52 | |||||
| MM/GBSA SP | 2ATH | A1 A20 A22 A27 A46 A53 A56 A58 A59 | 2ATH | B7 B26 B37 | 2ATH | C3 C10 C25 C34 C35 |
| 2I4J | A9 A11 A12 A13 A14 A16 A23 A24 A37 A39 A60 | 2F4B | B1 B2 B4 B21 B36 | 2PRG | C5 C8 C11 C13 C15 C16 C19 C21 C22 C26 C36 C37 C42 C46 C50 | |
| 2PRG | A2 A29 A40 | 2PRG | B16 B23 B24 B27 B28 B29 B31 B32 B38 | 2Q59 | C1 C2 C7 C27 C29 C31 C38 C44 | |
| 2VV0 | A8 A10 A30 A31 A41 A42 A44 A51 A55 | 2VV1 | B14 B17 B18 B19 B22 B34 B35 B39 | 2XKW | C4 C6 C9 C17 C41 C47 | |
| 2VV2 | A7 A15 A17 A18 A19 A21 A26 A43 A50 | 2VV2 | B9 B11 B25 B30 | 3B3K | C24 C32 C40 C43 C48 | |
| 2XKW | A3 A6 A34 A36 A52 | 3CDS | B3 B6 B8 B15 | 3GBK | C14 C39 C45 | |
| 3B3K | A48 | 3HO0 | B10 B20 | 3HO0 | C33 | |
| 3GBK | A4 A5 A25 A28 A32 A33 A35 A38 A49 A54 | 3NOA | B5 B12 B13 B33 B40 | 3HOD | C23 C49 | |
| 3HO0 | A47 | 3NOA | C12 C20 C28 C30 | |||
| 3NOA | A45 A57 | 3QT0 | C18 | |||
| MM/GBSA XP | 2F4B | A1 A18 A19 A36 | 2I4J | B1 B6 B20 B26 | 2F4B | C8 C24 C25 C37 C40 |
| 2I4J | A3 A12 A13 A15 A16 A32 A34 A48 A50 | 2PRG | B16 B19 B23 B24 B25 B27 B28 B29 B30 B31 B32 B38 | 2I4J | C3 C6 C9 C17 C42 | |
| 2PRG | A28 A31 A40 A49 A59 | 2Q59 | B13 B15 B18 B21 B22 B35 B36 B40 | 2PRG | C1 C2 C10 C11 C14 C19 C20 C21 C22 C26 C27 C29 | |
| 2Q59 | A5 A7 A9 A10 A11 A17 A21 A23 A25 A26 A27 A37 A43 A47 A60 | 2VV1 | B14 B17 B33 B34 | 2VV1 | C7 C16 C44 C49 C50 | |
| 2VV0 | A14 A20 A22 A39 A41 A45 A51 A56 A57 | 2VV2 | B11 | 3CDS | C5 C13 C32 C43 C45 | |
| 2VV1 | A4 A29 A30 A38 A42 A44 A52 A55 | 3HOD | B2 B7 B8 B9 B10 B37 B39 | 3GBK | C41 C46 | |
| 2XKW | A2 A6 A8 A33 A35 A53 A54 | 3NOA | B3 B4 B5 B12 | 3HO0 | C28 C39 | |
| 3HOD | A58 | 3HOD | C18 C34 C48 | |||
| 3NOA | A24 A46 | 3NOA | C12 C15 C30 C31 C33 | |||
| 3QT0 | C4 C23 C35 C36 C38 C47 | |||||
In the case of Set 1, R2 had a good value when Glide SP docking algorithm was used (R2 = 0.63); and Glide XP did not surpass the SP mode. The best model was obtained when MM/GBSA re-scoring was applied to the poses obtained using Glide XP (R2 = 0.66, Fig. 3a and b). In any case, we achieved a good correlation in all cases (R2 = 0.56–0.66), indicating that all the methods capture the molecular features important for the binding of this compound series.
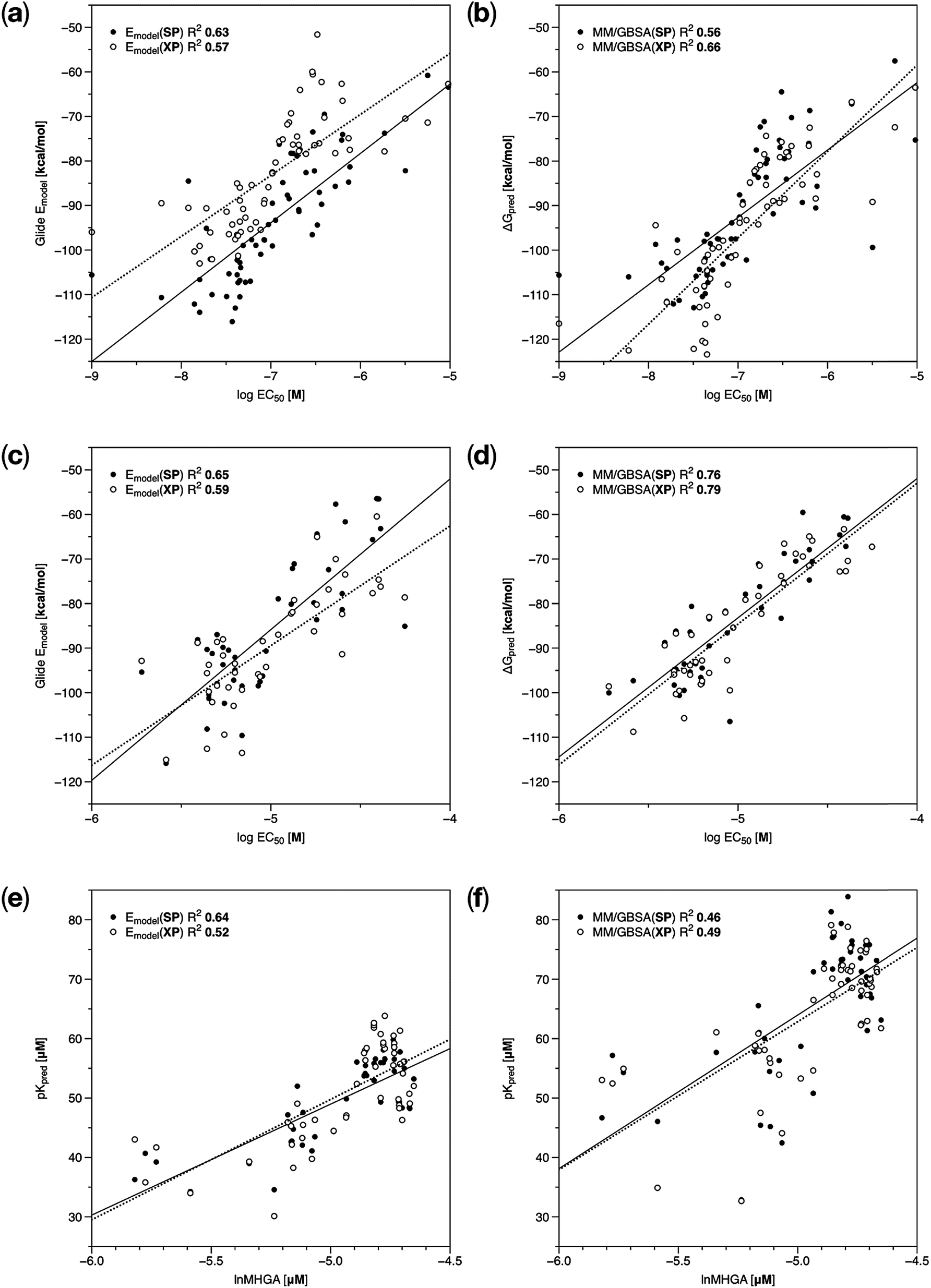 | ||
| Fig. 3 Regression plots of the experimental activities versus predicted activities for the developed models. (a) Estimated Glide scores for Set 1, (b) MM/GBSA re-scoring for Set 1, (c) estimated Glide scores for Set 2, (d) MM/GBSA re-scoring for Set 2, (e) estimated Glide scores for Set 3, (f) MM/GBSA re-scoring for Set 3. Filled circles represent SP mode and empty circles represent XP mode for each case. Trend lines for the results obtained from SP and XP modes are shown in solid and dotted lines, respectively. | ||
In the case of Set 2, MM/GBSA re-scoring produced a substantial improvement of the ligand ranking (R2 = 0.79, Table 4, Fig. 3c and d). This result suggests that protein flexibility plays a major role in the binding of these ligands. It is noteworthy that the substituents R1 (compounds B1–B40 in Fig. 1 and Table 2) in these ligands participate in the formation of HBs with the tetrad of aromatic residues previously defined as the ECIDALs. Therefore, it could be inferred that the minimization of residues surrounding the docked ligand structures during the refinement stage of the MM/GBSA protocol was able to enhance some HB and hydrophobic interactions in the binding interface, where the Glide docking approach was not able to model these effects properly.
In the case of Set 3, the best performance was obtained for Glide SP method (R2 = 0.64, Table 4, Fig. 3e and f). The more rigorous Glide XP docking algorithm deteriorates the results obtained using the SP mode. In addition, MM/GBSA re-scoring performed worse than the docking approach, and in fact, it was not capable to provide a suitable ranking.
Binding modes of PPARγ agonists
PPARγ binding site is larger than those found in other nuclear receptors. It is delimited by the helices H2′, H3, H4, H5, H7, H10/H11, H12 (the activating function-2 or AF-2), and the β-strands S3 and S4. The binding site has three pockets with a global “Y”-shape.37 The first one is the entrance pocket, which extends from the surface of the protein. It has polar residues at the solvent accessible part, and hydrophobic residues at the inner part; it branches off to the pockets arm I and arm II. Arm I is a polar cavity extending toward the H12 helix (AF-2); it contains the ECIDALs defined above. Finally, arm II is located between H3 and S3/S4, it is mainly hydrophobic.The PPARγ-agonists structures deposited in PDB depict the typical positions of the polar head and a hydrophobic tail of the active ligands. The known crystallographic 3D structures have revealed that the ligand polar heads, such as the carboxylate group21,22 or TZD group,38–40 form a HB network with the tetrad of aromatic residues defined as the ECIDALs in our study, located at the arm I; meanwhile, the ligand hydrophobic tails occupy the hydrophobic arm II, and/or the hydrophobic part of the entrance pocket. The analysis of crystallographic 3D structures also gives an important information about the induced fits in the binding pockets upon ligand binding, i.e. conformational changes of the side chain of the residues allowing a better binding of the ligand. For instance, the residues Phe282, Arg288, Phe363, and Tyr473 adopt different conformations among the available different crystallographic structures.41–43 For this reason, it is reasonable to consider the flexibility of these residues in docking experiments. Given the availability of several structures of the PPARy ligand binding domain co-crystallized with agonists, cross-docking experiments were performed to take into account these induced-fit effects upon ligand binding by carrying out docking calculations on multiple receptor conformations, as a practical alternative to incorporate protein flexibility.44 Cross-docking method proved to be a useful strategy for structure-based drug design in previous reports.45–47
The poses obtained for each set occupy the above mentioned PPARγ pockets. The poses that are included in the best models according to global R2 values for the three studied sets (represented with bold letters in Table 4) are represented in the Fig. 4. All binding poses displayed favorable interactions with the ECIDALs; HB interactions were identified between the studied ligands and at least one of the polar aromatic residues located in arm I. Table 6 shows the main HB interactions with these residues observed for the compounds in Sets 1–3 as PPARγ agonists. In general, the carboxylate head groups of compounds from Set 1 form strong HBs with the side chain groups of Tyr473 (H12), Tyr327 (H5), His323 (H4), His449 (H11), and Ser289 (H3) with distances between heavy atoms around 3.0 Å (Table 6). These interactions are also important for Set 2, but the interactions with Tyr327 and Tyr473 seem to be less important (Table 6). On the other hand, TZD groups of the compounds from Set 3 have HB interactions mainly with Ser289 and His323, and have no HB interactions with His449.
 | ||
| Fig. 4 Binding modes for compounds from Set 1 (A), Set 2 (B), and Set 3 (C) obtained by using the best models highlighted in Table 4. Ligand structures are shown in different colors according to the occupancy of the ligand hydrophobic tails. Compounds in green occupy arm II, compounds in yellow occupy the entrance hydrophobic part, and compounds in purple occupy both pockets. Compounds in cyan occupy the deeper part of the arm I. | ||
| Residues | Set 1 | Set 2 | Set 3 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| %b | d (Å) | αacc (°)c | αdonor (°)c | %b | d (Å) | αacc (°)c | αdonor (°)c | %b | d (Å) | αacc (°)c | αdonor (°)c | |
| a Interactions with a relative frequency less than 10% are omitted.b % represents relative interaction frequency, where 100% indicates that it is established by all molecules within the corresponding ligand set.c αacc and αdonor indicate the acceptor (H⋯A–R) and donor (D–H⋯A) angles, respectively. | ||||||||||||
| Ser289 (HB) | 43% | 2.82 ± 0.06 | 133.5 ± 22.5 | 160.4 ± 7.6 | 45% | 2.93 ± 0.13 | 136.8 ± 25.3 | 150.3 ± 10.3 | 42% | 2.90 ± 0.14 | 116.2 ± 18.7 | 159.4 ± 12.8 |
| His323 (HB) | 42% | 3.07 ± 0.14 | 115.2 ± 12.2 | 158.7 ± 15.2 | 38% | 2.95 ± 0.10 | 132.5 ± 17.3 | 145.8 ± 14.5 | 38% | 2.95 ± 0.15 | 147.4 ± 7.7 | 142.2 ± 9.4 |
| Tyr327 (HB) | 43% | 3.02 ± 0.19 | 144.9 ± 23.5 | 161.5 ± 10.5 | 30% | 2.89 ± 0.08 | 124.4 ± 15.5 | 168.3 ± 5.0 | 14% | 2.95 ± 0.12 | 115.7 ± 20.1 | 149.8 ± 11.9 |
| His449 (HB) | 48% | 2.85 ± 0.09 | 121.8 ± 13.7 | 157.2 ± 15.2 | 50% | 2.97 ± 0.09 | 114.9 ± 13.9 | 150.1 ± 14.7 | ||||
| Tyr473 (HB) | 53% | 2.98 ± 0.18 | 113.1 ± 15.6 | 146.5 ± 12.8 | 38% | 2.83 ± 0.07 | 127.4 ± 26.7 | 155.9 ± 11.5 | 18% | 3.21 ± 0.23 | 106.6 ± 10.1 | 145.0 ± 5.5 |
In all the sets the polar head groups are located inside the arm I, while the hydrophobic tail formed diverse interactions with different residues located in arm II and the hydrophobic part of the entrance pocket (Fig. 4). The majority of compounds from the Set 3 have the hydrophobic tail inside the arm II (green compounds in Fig. 4C). Compounds from the sets 1 and 2 have the hydrophobic tail inside arm II or the entrance in a similar proportion, but several compounds from the Set 2 occupy both pockets at the same time (purple compounds in Fig. 4B). Some compounds from the Set 3 extend to the deeper part of the arm I (cyan compounds). There are no evidences of PPARγ agonist structures in the available crystallographic data with a binding position in this zone; therefore, we consider that these conformations are no reliable solutions of the docking search.
The models developed in this work were constructed by gathering sixteen crystallographic structures to represent varying conformations of the side-chains in the protein's active site. A question that comes to mind is: why several crystallographic structures were needed to create successful models? We have to consider that residues adopt different conformations among the available crystallographic structures; some of them, such as Phe282, Phe363, Tyr473 (arm I), and Arg288 (entrance) have completely different orientations in the available crystallographic structures, which cause significant differences in the global shape of PPARγ binding sites.41–43 Analysis of the conformational differences observed for these residues are reported in ESI (Fig. S2–S5†). Fig. S2† shows root mean square deviation (RMSD) values for comparison between these residues in the sixteen crystallographic structures used in our protocol. We observed that Phe282 has two orientations (orientations I and II), Arg288 has two orientations (I and II), Phe363 has three orientations (I, II, and III), and Tyr473 has minor changes but has three orientations (I, II, and III). When only one crystallographic study is considered for docking, we are ignoring that ligands can induce large conformational changes in the selected residues; therefore, the use of several structures seems to be a more rational approach.
Fig. S3† shows the conformational changes of the residues Phe282, Arg288, Phe363, and Tyr473 in the protein structures used to build the best correlation models (Tables 4 and 5). For Set 1, Phe282 and Arg288 have large conformational changes only in one of the nine crystallographic structures used to develop the best model (structure with PDB code 3HOD), and this structure was used only to dock one compound from Set 1 according to Table 5 (compound A58 in MM/GBSA re-scoring from Glide XP). In addition, Tyr473 has large conformational changes only in the structure with PDB code 3NOA, and this structure was used only to dock two compounds from Set 1 according to Table 5 (compounds A24 and A46 in MM/GBSA re-scoring from Glide XP). Therefore, the best model for Set 1 included mainly large changes in Phe363. For Set 2, Phe282, Arg288, and Tyr473 have also large conformational changes only in the crystallographic structures 3HOD and 3NOA, but eleven compounds from Set 2 were docked in these structures (Table 5); therefore, the best model included large changes in the four residues. Finally, there are large changes in the four residues in the structures used to build the best correlation model for Set 3 (Fig. S3c†).
It is noteworthy that arm I residues Phe282 (H3) and Phe363 (H7) are very close; therefore, combination of the orientations I and II of Phe282 and orientations I, II, and III of Phe363 lead to four conformational arrangements: F282(I)F363(I), F282(I)F363(II), F282(I)F363(III), and F282(II)F363(I). Fig. S4† shows root mean square deviation (RMSD) values for comparison between the Phe282–Phe363 pair in the sixteen crystallographic structures used in our protocol. In addition, Fig. S5† shows the conformational changes of the Phe282–Phe363 pair in the structures used to build the best correlation models (Tables 4 and 5). There are large changes in this pairs in the structures used to build the best correlation models for the studied sets. These changes signify changes in volume and shape of the binding site in this zone, which has an influence in the ligands binding to PPARγ.
Conclusion
A cross-docking protocol, using docking algorithms Glide SP and XP considering their scoring functions, and MM/GBSA re-scoring method, was used to model the structure–activity relationship of three series of chemically-diverse PPARγ agonists named Set 1, 2 and 3 in this manuscript.The studied compounds were docked against sixteen PPARγ crystallographic structures. Some considerations were addressed during posing and scoring/ranking processes. Conformations that comply with ECIDALs were privileged during posing process; i.e., poses which fit within a target protein's binding site and have the chemical interactions previously observed for the known crystallized PPARγ ligands were selected. On the other hand, the scoring/ranking process was performed by optimizing the correlation (R2 value) between the calculated score values and the experimental activities; i.e., for each studied compound one ligand–PPARγ complex obtained using docking was selected (from the pool of sixteen complexes) to get the highest R2 value.
The proposed methodology was successful in most of the cases, since high R2 correlation values between the calculated energies and the logarithmic activities were obtained for the studied sets. The inclusion of protein flexibility through the use of several crystal receptor conformations was necessary to meet our goals. Such strategy was used to consider ligand-induced fit effects in the binding site.
In general, the employed methodology serves as a valid approach to study PPARγ ligands with computational methods. It provides a description of the interactions between PPARγ and its agonists, and it may be a practical approach for the design and computational evaluation of more potent candidates.
Acknowledgements
This work is supported by Fondecyt # 1130216 (I. P. and J. C.) from CONICYT, Chile. E. F. acknowledges support from Fondecyt # 11140142. C. M. G. and F. A. C. acknowledge support from doctoral fellowships CONICYT-PCHA/Folio 21120214 and 21120213, respectively.References
- V. Fuster, B. B. Kelly and R. Vedanthan, J. Am. Coll. Cardiol., 2011, 58, 1208–1210 CrossRef PubMed.
- D. A. Vorchheimer and R. Becker, Mayo Clin. Proc., 2006, 81, 59–68 CrossRef CAS PubMed.
- V. Fuster, L. Badimon, J. J. Badimon and J. H. Chesebro, N. Engl. J. Med., 1992, 326, 310–318 CrossRef CAS PubMed.
- E. Fuentes, F. Fuentes, V. Andrés, O. M. Pello, J. Font de Mora and I. Palomo, Platelets, 2013, 24, 255–262 CrossRef PubMed.
- Z. M. Ruggeri, Thromb. Haemostasis, 1997, 78, 611–616 CAS.
- M. Gawaz, H. Langer and A. E. May, J. Clin. Invest., 2005, 115, 3378–3384 CrossRef CAS PubMed.
- L. A. Moraes, L. Piqueras and D. Bishop-Bailey, Pharmacol. Ther., 2006, 110, 371–385 CrossRef CAS PubMed.
- D. Li, K. Chen, N. Sinha, X. Zhang, Y. Wang, A. K. Sinha, F. Romeo and J. L. Mehta, Cardiovasc. Res., 2005, 65, 907–912 CrossRef CAS PubMed.
- W. Ahmed, O. Ziouzenkova, J. Brown, P. Devchand, S. Francis, M. Kadakia, T. Kanda, G. Orasanu, M. Sharlach, F. Zandbergen and J. Plutzky, J. Intern. Med., 2007, 262, 184–198 CrossRef CAS PubMed.
- M. Lehrke and M. A. Lazar, Cell, 2005, 123, 993–999 CrossRef CAS PubMed.
- C. Duval, G. Chinetti, F. Trottein, J.-C. Fruchart and B. Staels, Trends Mol. Med., 2002, 8, 422–430 CrossRef CAS PubMed.
- D. S. Sim, G. Merrill-Skoloff, B. C. Furie, B. Furie and R. Flaumenhaft, Blood, 2004, 103, 2127–2134 CrossRef CAS PubMed.
- F. Akbiyik, D. M. Ray, K. F. Gettings, N. Blumberg, C. W. Francis and R. P. Phipps, Blood, 2004, 104, 1361–1368 CrossRef CAS PubMed.
- H. Du, H. Hu, H. Zheng, J. Hao, J. Yang and W. Cui, Thromb. Res., 2014, 134, 111–120 CrossRef CAS PubMed.
- F. Y. Ali, S. J. Davidson, L. A. Moraes, S. L. Traves, M. Paul-Clark, D. Bishop-Bailey, T. D. Warner and J. A. Mitchell, FASEB J., 2006, 20, 326–328 CAS.
- N. T. Dickinson, E. K. Jang and R. J. Haslam, Biochem. J., 1997, 323(2), 371–377 CrossRef CAS PubMed.
- T. Myles, T. Nishimura, T. H. Yun, M. Nagashima, J. Morser, A. J. Patterson, R. G. Pearl and L. L. K. Leung, J. Biol. Chem., 2003, 278, 51059–51067 CrossRef CAS PubMed.
- A. Farce, N. Renault and P. Chavatte, Curr. Med. Chem., 2009, 16, 1768–1789 CrossRef CAS PubMed.
- X. Y. Xu, F. Cheng, J. H. Shen, X. M. Luo, L. L. Chen, L. D. Yue, Y. Du, F. Ye, S. H. Jiang, D. Y. Zhu, H. L. Jiang and K. X. Chen, Int. J. Quantum Chem., 2003, 93, 405–410 CrossRef CAS.
- K. W. Nettles, Nat. Struct. Mol. Biol., 2008, 15, 893–895 CAS.
- D. B. Lowe, N. Bifulco, W. H. Bullock, T. Claus, P. Coish, M. Dai, F. E. Dela Cruz, D. Dickson, D. Fan, H. Hoover-Litty, T. Li, X. Ma, G. Mannelly, M.-K. Monahan, I. Muegge, S. O'Connor, M. Rodriguez, T. Shelekhin, A. Stolle, L. Sweet, M. Wang, Y. Wang, C. Zhang, H.-J. Zhang, M. Zhang, K. Zhao, Q. Zhao, J. Zhu, L. Zhu and M. Tsutsumi, Bioorg. Med. Chem. Lett., 2006, 16, 297–301 CrossRef CAS PubMed.
- D. Merk, C. Lamers, J. Weber, D. Flesch, M. Gabler, E. Proschak and M. Schubert-Zsilavecz, Bioorg. Med. Chem., 2015, 23, 499–514 CrossRef CAS PubMed.
- T. Sohda, K. Mizuno, E. Imamiya, Y. Sugiyama, T. Fujita and Y. Kawamatsu, Chem. Pharm. Bull., 1982, 30, 3580–3600 CrossRef CAS PubMed.
- B. R. P. Kumar, S. Sopna, J. Verghese, B. Desai and M. J. Nanjan, Med. Chem. Res., 2011, 21, 624–633 CrossRef.
- H. E. Xu, M. H. Lambert, V. G. Montana, K. D. Plunket, L. B. Moore, J. L. Collins, J. A. Oplinger, S. A. Kliewer, R. T. Gampe, D. D. McKee, J. T. Moore and T. M. Willson, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 13919–13924 CrossRef CAS PubMed.
- J. M. Lehmann, L. B. Moore, T. A. Smith-Oliver, W. O. Wilkison, T. M. Willson and S. A. Kliewer, J. Biol. Chem., 1995, 270, 12953–12956 CrossRef CAS PubMed.
- J. L. Banks, H. S. Beard, Y. Cao, A. E. Cho, W. Damm, R. Farid, A. K. Felts, T. A. Halgren, D. T. Mainz, J. R. Maple, R. Murphy, D. M. Philipp, M. P. Repasky, L. Y. Zhang, B. J. Berne, R. A. Friesner, E. Gallicchio and R. M. Levy, J. Comput. Chem., 2005, 26, 1752–1780 CrossRef CAS PubMed.
- R. A. Friesner, R. B. Murphy, M. P. Repasky, L. L. Frye, J. R. Greenwood, T. A. Halgren, P. C. Sanschagrin and D. T. Mainz, J. Med. Chem., 2006, 49, 6177–6196 CrossRef CAS PubMed.
- M. D. Eldridge, C. W. Murray, T. R. Auton, G. V. Paolini and R. P. Mee, J. Comput.–Aided Mol. Des., 1997, 11, 425–445 CrossRef CAS PubMed.
- R. A. Friesner, J. L. Banks, R. B. Murphy, T. A. Halgren, J. J. Klicic, D. T. Mainz, M. P. Repasky, E. H. Knoll, M. Shelley, J. K. Perry, D. E. Shaw, P. Francis and P. S. Shenkin, J. Med. Chem., 2004, 47, 1739–1749 CrossRef CAS PubMed.
- J. Li, R. Abel, K. Zhu, Y. Cao, S. Zhao and R. A. Friesner, Proteins, 2011, 79, 2794–2812 CrossRef CAS PubMed.
- J. Caballero, C. Muñoz, J. H. Alzate-Morales, S. Cunha, L. Gano, R. Bergmann, J. Steinbach and T. Kniess, Eur. J. Med. Chem., 2012, 58, 272–280 CrossRef CAS PubMed.
- C. Munoz, F. Adasme, J. H. Alzate-Morales, A. Vergara-Jaque, T. Kniess and J. Caballero, J. Mol. Graphics Modell., 2012, 32, 39–48 CrossRef CAS PubMed.
- C. Mulakala and V. N. Viswanadhan, J. Mol. Graphics Modell., 2013, 46, 41–51 CrossRef CAS PubMed.
- F. Adasme-Carreño, C. Muñoz-Gutierrez, J. Caballero and J. Alzate-Morales, Phys. Chem. Chem. Phys., 2014, 16, 14047–14058 RSC.
- C. Navarro-Retamal, C. Gaete-Eastman, R. Herrera, J. Caballero and J. H. Alzate-Morales, PLoS One, 2016, 11, e0153057 Search PubMed.
- V. Zoete, A. Grosdidier and O. Michielin, Biochim. Biophys. Acta, 2007, 1771, 915–925 CrossRef CAS PubMed.
- R. T. Nolte, G. B. Wisely, S. Westin, J. E. Cobb, M. H. Lambert, R. Kurokawa, M. G. Rosenfeld, T. M. Willson, C. K. Glass and M. V. Milburn, Nature, 1998, 395, 137–143 CrossRef CAS PubMed.
- H. Hauner, Diabetes/Metab. Res. Rev., 2002, 18(2), S10–S15 CrossRef CAS PubMed.
- R. Dumasia, K. A. Eagle, E. Kline-Rogers, N. May, L. Cho and D. Mukherjee, Curr. Drug Targets: Cardiovasc. & Haematol. Disord., 2005, 5, 377–386 CAS.
- I.-L. Lu, C.-F. Huang, Y.-H. Peng, Y.-T. Lin, H.-P. Hsieh, C.-T. Chen, T.-W. Lien, H.-J. Lee, N. Mahindroo, E. Prakash, A. Yueh, H.-Y. Chen, C. M. V. Goparaju, X. Chen, C.-C. Liao, Y.-S. Chao, J. T.-A. Hsu and S.-Y. Wu, J. Med. Chem., 2006, 49, 2703–2712 CrossRef CAS PubMed.
- S. Ebdrup, I. Pettersson, H. B. Rasmussen, H.-J. Deussen, A. Frost Jensen, S. B. Mortensen, J. Fleckner, L. Pridal, L. Nygaard and P. Sauerberg, J. Med. Chem., 2003, 46, 1306–1317 CrossRef CAS PubMed.
- E. Burgermeister, A. Schnoebelen, A. Flament, J. Benz, M. Stihle, B. Gsell, A. Rufer, A. Ruf, B. Kuhn, H. P. Märki, J. Mizrahi, E. Sebokova, E. Niesor and M. Meyer, Mol. Endocrinol., 2006, 20, 809–830 CrossRef CAS PubMed.
- M. Totrov and R. Abagyan, Curr. Opin. Struct. Biol., 2008, 18, 178–184 CrossRef CAS PubMed.
- R. Ragno, S. Frasca, F. Manetti, A. Brizzi and S. Massa, J. Med. Chem., 2005, 48, 200–212 CrossRef CAS PubMed.
- A. May and M. Zacharias, J. Med. Chem., 2008, 51, 3499–3506 CrossRef CAS PubMed.
- T. Tuccinardi, G. Poli, V. Romboli, A. Giordano and A. Martinelli, J. Chem. Inf. Model., 2014, 54, 2980–2986 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra12084a |
| This journal is © The Royal Society of Chemistry 2016 |