
Monodisperse magnetite nanoparticles with nearly ideal saturation magnetization†
Scott J. Kempa,
R. Matthew Ferguson *a,
Amit P. Khandhara and
Kannan M. Krishnanab
*a,
Amit P. Khandhara and
Kannan M. Krishnanab
aLodeSpin Labs, 225 Fluke Hall, 4000 Mason Road, Seattle, Washington 98195, USA. E-mail: matt@lodespin.com
bDepartment of Materials Sciences & Engineering, Roberts Hall, University of Washington, Seattle, Washington 98195, USA
First published on 8th August 2016
Abstract
We present a scalable thermolysis and high temperature oxidation procedure for synthesizing monodisperse magnetite nanoparticles with saturation magnetization of up to 80 emu g−1 (412 kA m−1), 92% of bulk magnetite. Diameters in the 15–30 nm size range are produced from iron oleate via the thermolysis method at 324 °C and varying oleic acid ratios for size control (6.7–7.6 equivalents per Fe). The influence of the iron oleate synthesis procedure on the quality of resulting nanoparticles is examined and the structure of the iron oleate is proposed to have a triironoxonium core [Fe3O+] based on magnetic susceptibility measurements. The thermolysis method is shown to initially give wüstite nanoparticles, which are oxidized in situ at 318 °C using 1% oxygen in argon to form highly magnetic magnetite nanoparticles. The use of 1% oxygen offers broad application as a safe and efficient reagent for the high temperature oxidation of nanoparticles. Special consideration to the reproducibility of nanoparticle diameter and monodispersity has uncovered critical factors. Additionally, the reduction of Fe(III) to Fe(II) is shown to occur during the heat up stage of thermolysis, beginning at less than 180 °C and being complete by 320 °C. Evidence for the reduction occurring by the oxidative decarboxylation of oleic acid is presented. Decomposition of the remaining oleic acid is shown to occur by a ketonization reaction producing oleone. The nucleation event and growth of particles is examined by TEM. Comparison of the solvents 1-octadecene and octadecane are presented demonstrating the effect on the reduction of Fe(III) during heat up, the large difference in particle size, and effects on the oxidation rate of iron oxide nanoparticles. Determination of Fe(II) content in magnetic iron oxide nanoparticles by titration is presented.
1. Introduction
Magnetic iron oxide nanoparticles have proven useful in a wide range of applications, such as ferrofluids, magnetic separations, and medicine.1,2 In the clinic, iron oxides are used as magnetic resonance imaging (MRI) contrast agents and for treating iron deficiency, while emerging applications include drug delivery, hyperthermia3 and magnetic particle imaging (MPI).4,5 Iron oxide nanoparticles are also a fundamental component in the study of magnetism.6,7Several approaches for producing iron oxide nanoparticles have been developed, generally classifiable as water-based co-precipitation reactions, or non-polar solvent-based thermolysis reactions. Thermolysis produces monodisperse nanoparticles with controllable size, and therefore tunable physical properties, which is very attractive for applications research. Understandably, thermolysis reactions have been reported using a variety of different iron precursors, including iron pentacarbonyl by Hyeon,8 Fe(acac)3 by Sun,9,10 iron oxyhydroxide by Colvin,11 and iron oleate by Peng12 and Hyeon.13 Thermolysis typically uses an iron oleate (or other iron–organic precursor) mixed with octadecene (or other high boiling solvent) and oleic acid (or other surfactant) and heated to high temperature (250 to 360 °C), at which point formation of iron oxide nanoparticles occurs. Depending on the solvent, surfactant and temperature, a range of different sizes can be obtained with a dispersity of 5–10%.
While size control has been extensively studied and reported,14 in many cases no direct control of iron oxidation state is implemented during synthesis. This can result in observed saturation magnetization below bulk magnetite (86 emu g−1; 446 kA m−1).13 Iron oxide nanoparticles can exist in numerous phases with very different magnetic properties, including wüstite, magnetite, and maghemite. Magnetite has the highest magnetic saturation, which makes it desirable for many applications. Smaller particles (<10 nm) are commonly found to be maghemite (Fe2O3) and larger particle (>15 nm) are commonly found to be magnetite. In many cases, the nanoparticles can consist of a mixture of phases such as maghemite/magnetite (Fe3−xO4) or wüstite core with a magnetite shell. Proper control of oxidation state or the lack thereof has not yet been properly addressed, leading to high variability in the magnetic properties of iron oxide nanoparticles synthesized using similar procedures.
Herein, we report the development of a scalable procedure for synthesizing magnetite nanoparticles with diameter ranging from 15 to about 35 nm, resulting from over 200 nanoparticle synthesis reactions. With this procedure, described generally in Scheme 1, nanoparticle size is controlled by varying an excess of oleic acid while keeping the reaction temperature at 324 °C. We focus sequentially on three critical stages of nanoparticle synthesis: first, we discuss synthesis and the structure of iron(III) oleate and its influence on nanoparticle formation. Next, the thermolysis reaction is discussed, including several controls introduced to improve the reproducibility of nanoparticle formation during thermolysis. Finally, since nanoparticles form as the wüstite phase, to achieve the desired magnetite phase the use of 1% oxygen in argon as an efficient and safe oxidant of iron oxide nanoparticles is introduced. Proper execution of these procedures produces high quality magnetite particles (see Fig. 1). We have made a particular effort to reference the extensive literature on iron oxide nanoparticle synthesis so this manuscript can be a reference to those wishing to produce nanoparticles for applications research and for those interested in further study of this important, but imperfectly understood reaction.
 | ||
| Scheme 1 Magnetite nanoparticle via thermolysis. | ||
 | ||
| Fig. 1 Magnetite nanoparticles synthesized by thermolysis with oxidation in 1% oxygen in argon. (a) Bright field TEM image, (b) selected area electron diffraction reveals spinel iron oxide. (c) HR TEM shows highly crystalline particles. | ||
2. Experimental
2.1. Materials
1-Octadecene (tech. 90%), oleic acid (tech. 90%), and iron trichloride hexahydrate (ACS, 97.0–102.0%) were obtained from Alfa Aesar. Sodium oleate (>97%) was obtained from Tokyo Chemical Industry CO, LTD. 1% oxygen in argon was obtained from Praxair. Water used in any experiment was purified at 18.2 Mohm cm. Poly(maleic anhydride-alt-1-octadecene) (average Mn 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–50000 Da) was obtained from Sigma-Aldrich. mPEG-NH2 (MW = 20000 g mol−1) was purchased from JenKem. Infrared spectra were obtained using a Bruker Vertex 70 FTIR with ATR.
000–50000 Da) was obtained from Sigma-Aldrich. mPEG-NH2 (MW = 20000 g mol−1) was purchased from JenKem. Infrared spectra were obtained using a Bruker Vertex 70 FTIR with ATR.
2.2. Synthesis of iron(III) oleate
This is a modification of a published procedure.13 To a 2 liter three neck round bottom flask equipped with a 1-1/2 × 5/8 inch Teflon coated magnetic stir bar, was added sodium oleate (147.05 g, 483 mmol) and hexanes (500 mL). The flask was equipped with a glass stopper in the left neck, a SUBA-SEAL® septum with a thermocouple in the right neck, and reflux condenser topped with a Schlenk line attachment on the center neck. The mixture was stirred to suspend the sodium oleate, then ethanol (300 mL) was added. The slow (30 seconds) addition of water (60 mL) caused nearly all of the solids to dissolve. The reaction vessel was equipped with a heating mantle and heated to 40 °C with stirring, at which point the sodium oleate had completely dissolved. A solution of iron(III) trichloride hexahydrate (43.518 g, 161 mmol) in water (100 mL) was prepared in a 250 mL Erlenmeyer flask with stirring for about 30 minutes, at which time the iron(III) chloride had completely dissolved. The iron(III) chloride solution was added to the reaction vessel via a funnel with pre-wetted qualitative filter paper (15 cm) and washed in with water (20 mL). The reaction vessel was purged with argon for 1 minute and then heated to a gentle reflux (57 °C internal temperature). The reaction was held at reflux and stirring (500 rpm) was maintained for 4 hours. The heating mantle was then removed and the reaction was allowed to cool to 50 °C, then transferred to a 1 liter separatory funnel. The bottom layer was drained and the upper red layer was washed with water (3 × 150 mL, 10 second shake period). The dark red organic layer was then transferred to a 1 liter Erlenmeyer flask containing anhydrous sodium sulfate (50 g). The solution was swirled occasionally for 10 minutes and then filtered through qualitative filter paper into a 2 liter round bottom flask. The solution was concentrated carefully on a rotary evaporator using a water aspirator for vacuum, first at a water bath temperature of 20 °C and then increased in small increments to 30 °C. After solvent removal appeared to have ceased, the vacuum source was switched to high vacuum on the rotary evaporator and concentrating was continued for about 30 minutes at 30 °C bath temperature. After drying on a high vacuum line overnight, the resulting dark red syrup (144.05 g) was deemed to contain 160 mmol of iron(III) oleate and could be divide by mass for use in the nanoparticle synthesis. IR (neat, cm−1): 3600–2500w, br, 2922s, 2852s, 1711m, 1587m, 1564w, 1527m, 1438m, 723w, 613w, br, 514w. Elemental analysis of crude product: found (%): C, 69.56; H, 11.05; N, 0.02; Fe (by ICP), 5.97%. Calculated (%) for Fe3O(oleate)7·2oleic acid·4.5H2O: C, 69.50; H, 11.09; Fe, 5.98.2.3. Measurement of magnetic susceptibility of iron(III) oleate
Samples for magnetic susceptibility measurement were prepared by adding 100 mg of iron(III) oleate to a gelatin capsule. Magnetization curves were measured up to 1 T at room temperature (295 K) using a LakeShore VSM. Data was corrected by removing the contribution of the sample holder and diamagnetism of the samples estimated from Pascal's constants.15,162.4. Synthesis of magnetite iron oxide nanoparticles
Oleic acid to Fe ratio: 7.3:1. To a 1 liter 3-neck heavy walled round bottom flask with 24/40 joints was added iron(III) oleate (40 mmol, 36.00 g), followed by oleic acid (82.478 g, 304 mmol) and 1-octadecene (200 g). The flask was equipped with a 1-1/2 × 5/8 inch Teflon coated magnetic stir bar, a glass stopper in the center neck, a SUBA-SEAL® septum with a thermocouple in the right neck, and a bump trap topped with an air condenser and Schlenk line attachment on the left neck. A DigiTrol II attached to a heating mantle was used to control the heating of the reaction vessel. The glass joints were sealed with a few drops of 1-octadecene. The reaction mixture was heated to 50 °C, held under vacuum and stirred at 450 rpm for 18 hours. The reaction was evacuated and filled with argon five times (holding vacuum for 5 minutes each time) and then purged with argon for 5 minutes. The upper half of the reaction vessel and the necks were wrapped in foil to reduce water condensation. The set point was changed to 110 °C. After 15 minutes the internal temperature was 122 °C. The controller was set to ramp at 5 °C min−1 and the set point was changed to 318 °C. The stir rate was increased to 800 rpm. Purging with argon (∼40 mL min−1) was continued via a 16 G needle through the septum in the right flask neck to aid in the removal of water vapor into the bump trap. When the temperature reached 318 °C, the argon purge through the septum was stopped and the Schlenk line was used to maintain an atmosphere of argon. The argon purging line and needle were removed from the septum. Approximately 2 minutes later, the reaction temperature reached 324 °C. Over the next 30 minutes the set point was gradually increased in 2 °C increments to maintain the temperature at 324 °C. After 1 hour 40 minutes, since reaching 318 °C, the reaction mixture had darkened and finally turned turbid with the color of milk chocolate indicating particle formation. After an additional 30 minutes the set point was changed to 318 °C. When the reaction had cooled to 318 °C (about 15 minutes), the addition of 1% oxygen in argon was begun at a flow rate of approximately 140 mL min−1 via a 16 gauge × 6-inch stainless steel needle immersed about 1 inch into the reaction mixture. After 3 hours of 1% oxygen in argon addition the reaction had turned black. The needle was pulled up so the tip of the needle was about 2 inches above the surface of the reaction mixture and the flow rate of 1% oxygen was reduced to about 15 mL min−1. The stir rate was reduced to about 450 rpm to prevent possible loss of stirring during the night. The reaction was kept at 318 °C for 34 hours from the time the reaction first reached 318 °C (28.5 hours from the point the 1% oxygen flow was reduced). The heating was turned off using a timer and the reaction was allowed to cool during the night. The cooled reaction mixture had thicken and the reaction mixture was carefully warmed to liquefy the mixture. When the reaction mixture was at 50–60 °C the mixture was transfer to a 500 mL bottle with the aid of hexanes (100 mL) and purged with argon. The above procedure was repeated multiple times with varying ratios of oleic acid to Fe to provide batches of nanoparticles with a variety of core diameters. Representative batches are recorded in Table 2.
2.5. Iron oxide nanoparticle titration procedure for determining Fe(II) content
A washed and dried sample of iron oxide nanoparticles (∼60 mg) was gently and briefly swirled with concentrated HCl (1.8 mL) under argon. After resting for 10 minutes the mixture was sonicated for 5 minutes. The digestion was checked by observing any movement of the remaining black flakes to a strong magnetic field. When no movement was observed sonication was continued for an additional 5 minutes. The resulting HCl mixture was diluted by the addition of water (35 mL). A 0.05 N solution of potassium permanganate was freshly prepared. The titration was performed using a 1 mL Norm-Ject disposable syringe with a 25 gauge needle and the amount of KMnO4 solution delivered was determined by the mass of solution added (density of 0.05 N KMnO4 was determined during preparation to be 1.0018 g mL−1). The end point of the titration was reached when the light pale yellow solution changed to a light clear orange. The relative mass of oleic acid coating was determined by thermogravimetric analysis (TGA) (∼4%). The remaining mass (∼96%) was assumed to be Fe3O4 to calculate the total iron. Alternatively the total iron content could be determined by ICP analysis of the titrated solution and was found to be consistent with the TGA estimate. The Fe(II) content of the iron oxide nanoparticles is reported in Table 2.3. Results and discussion
3.1. Synthesis and structure of iron oleate
Iron oleate is a widely studied starting material for iron oxide nanoparticle thermolysis. Several methods of preparing iron oleate have been reported,17 including reaction of sodium oleate with iron(III) chloride at reflux13 or at room temperature,12 the reaction of oleic acid and iron oxyhydroxide,11 and the reaction of iron powder with oleic acid.18An often cited advantage of iron oleate for nanoparticle synthesis is that it can be synthesized in large quantities to provide a uniform precursor for large-scale reactions. While this is certainly an advantage, iron(III) oleate is also a complicated starting material; previous authors have reported, it can take different structures depending on how it is synthesized, and these variations can affect nanocrystal morphology.19 Other authors investigated ligand bonding within iron oleate to discuss observed formation of novel iron oxide 1D “nanowhiskers” at low growth temperatures.20 While it is apparent the structure of iron oleate plays an important role in the outcome of the thermolysis reaction; iron(III) oleate, as typically prepared for thermolysis, takes the form of a viscous liquid preventing structural determination by single crystal X-ray diffraction.
To better understand iron oleate, we thoroughly examined the literature pertaining to the structure of iron carboxylates. Many groups have reported iron(III) carboxylates contain a triironoxonium core,21,22 including some verified by X-ray crystallography.23,24 Chen et al. postulated this to be the structure of iron(III) oleate25 based on FTIR and elemental analysis. We support the conclusion iron(III) oleate forms a triironoxonium core, based on additional magnetic susceptibility analysis and our own FTIR measurements. We also verified the Fe(III) content by potassium permanganate titration, showing the absence of Fe(II).
It has been reported26 triironoxonium carboxylates have unique magnetic susceptibility (χp) due to the interactions of the three irons in the triironoxonium core. Knowing a reflux step is necessary to produce uniform nanoparticles (see ESI S1†) from previous studies of iron oleate prepared at room temperature only, we investigated the iron oleate evolution during synthesis by taking samples after 5, 45, and 240 minutes (standard preparation) at reflux and measuring their susceptibility. The χT product (room temperature) increased from 1.44 to 1.65 to 1.76 [cm3 K mol−1 (iron)] after 5, 45, and 240 minutes at reflux, respectively. For each sample, χT was much less than 4.35, the expected value of a single Fe(III) ion (S = 5/2, g = 2). Some representative literature values of paramagnetic susceptibility, χp, for trinuclear iron carboxylate susceptibilities are provided in Table 1. The similarity of our measured magnetic susceptibility with literature results for other triironoxonium cores strongly suggests the majority of the “as prepared” iron(III) oleate is the triironoxonium species as shown in Fig. 2.
| Core | Fe:oleic acid ratio |
Oxidation with 1% O2 (mmol of O2) | Core diameter – dN (nM) | Fe(II) by titration (%) | Ms | ||||
|---|---|---|---|---|---|---|---|---|---|
| 3 h | 28 h | Total | TEM (sigma) | VSM (sigma) | [kA m−1] | [emu g−1] | |||
| NP1 | 6.7 | 16 | 8 | 23 | 18.2 (0.06) | — | 19 | — | |
| NP2 | 7.4 | 13 | 11 | 24 | 20.5 (0.07) | 19.8 (0.02) | 21 | 412 | 80 |
| NP3 | 7.1 | 13 | 20 | 33 | 21.5 (0.08) | 22.7 (0.04) | 24 | 375 | 72 |
| NP4 | 7.2 | 16 | 20 | 36 | 21.9 (0.07) | 23.2 (0.04) | 22 | 370 | 71 |
| NP5 | 7.2 | 16 | 20 | 36 | 24.1 (0.07) | 24.4 (0.04) | 26 | 402 | 78 |
| NP6 | 7.3 | 16 | 20 | 36 | 24.4 (0.06) | 23.9 (0.05) | 26 | 395 | 76 |
| NP7 | 7.2 | 16 | 20 | 36 | 24.4 (0.09) | 24.2 (0.06) | 28 | 401 | 77 |
| NP8 | 7.2 | 16 | 20 | 36 | 25.3 (0.08) | 25.9 (0.04) | 26 | 377 | 73 |
| NP9 | 7.6 | 21 | 44 | 64 | 26.9 (0.06) | 25.8 (0.06) | 25 | 415 | 80 |
| NP10 | 7.5 | 16 | 8 | 23 | 26.2 (0.05) | 26.3 (0.04) | 25 | 375 | 72 |
| NP11 | 7.5 | 13 | 20 | 33 | 27.4 (0.07) | 27.5 (0.05) | 31 | 364 | 70 |
| NP12 | 7.4 | 16 | 20 | 36 | 27.4 (0.06) | 26.9 (0.03) | 27 | 372 | 72 |
| NP13 | 7.5 | 16 | 11 | 27 | 28.6 (0.08) | 26.7 (0.09) | 29 | 306 | 59 |
| NP14 | 7.5 | 16 | 11 | 27 | 29.2 (0.07) | 28.8 (0.06) | 30 | 345 | 67 |
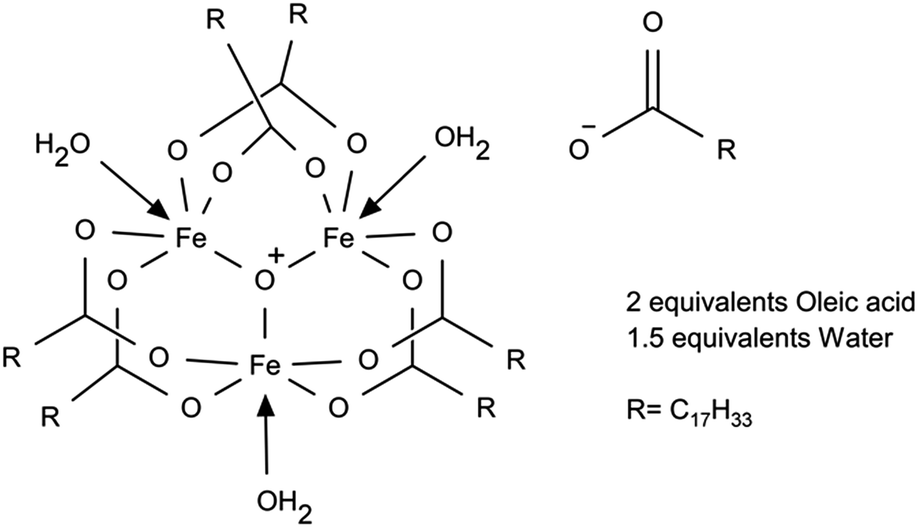 | ||
| Fig. 2 Proposed structure of iron oleate. | ||
The FT-IR spectrum of our iron(III) oleate (Fig. 3) was nearly identical to previously reported iron oleate prepared by Hyeon13 and very similar to unwashed iron oleate prepared by Bronstein.19 The key carboxylate region shows the following absorbances (cm−1): 1711m, 1587m, 1564w, 1527m, 1438m. The absorbance at 1711 cm−1 is due to free oleic acid. The absorbance at 1587 cm−1 correlates with the six oleates in a bridging formation (Δν = 149 cm−1). The absorbance at 1527 cm−1 correlates with the remaining non-bridging oleate binding in a bidentate fashion (Δν = 89 cm−1). The minor absorbance at 1564 cm−1 is not assigned, but is suspected of being a mononuclear core iron oleate or trace sodium oleate. A broad weak absorbance at 613 cm−1 is indicative of the Fe3O core and matches other triironoxonium cores reported in the literature.22
 | ||
| Fig. 3 FTIR of iron(III) oleate. | ||
Weight loss of iron III oleate during TGA (Fig. 4) was consistent with previous studies.13,20 Analysis of the weight loss events indicated loss of three oleates starting around 175 °C, followed by the loss of six oleates starting around 275 °C, which is consistent with six of the oleates being in the bridging formation. A possible alternative interpretation of the TGA analysis suggest the first loss of mass is due to oxidative decarboxylation of Fe(III) oleate to give Fe(II) oleate, followed by ketonization of the remaining oleates. Elemental analysis of as synthesized iron(III) oleate, showed a high correlation with [Fe3O(oleate)6(H2O)3][oleate](oleic acid)2(H2O)1.5 as depicted in Fig. 2. By measuring magnetic susceptibility, FTIR, and elemental analysis, the iron core structure and the mode of oleate binding of iron oleate can be monitored, even when structural determination by X-ray diffraction is not possible. The routine use of these three methods could provide valuable insight into the structure of iron(III) oleates made by various methods and could also explain the wide variations observed in the properties of nanoparticles synthesized by thermolysis.
 | ||
| Fig. 4 TGA (% wt loss and Δ% wt loss) of iron oleate compound. | ||
Finally, we note iron(III) oleate contains incorporated water and/or ethanol and oleic acid (either free or as ligands) and these components, and therefore the iron oleate structure, can fluctuate based on small changes in the procedure. For example, when the iron oleate solution in hexane was dried with sodium sulfate, extending the drying time from 10 to 45 minutes produced a thickened material that could no longer be gravity filtered (see ESI material S1† for examples of how nanoparticle morphology varied with iron oleate synthesis). As will be discussed in the next section, water also plays an important, but easily overlooked role in the thermolysis reaction.
3.2. Nanoparticle formation: thermolysis of iron oleate
Nanoparticle formation occurs during the thermolysis reaction, typically after heating the reaction mixture to 324 °C for an hour or more in the presence of excess oleic acid. The reaction's complexity is veiled by the simple procedure, which involves little more than setting a final temperature and ramp rate. We investigated the mechanism of particle formation and growth and studied reaction conditions to determine a thermolysis procedure that consistently yields high-quality particles. Here we present and discuss several significant findings related to: (1) nucleation and growth of iron oxide nanocrystals, (2) reduction of iron(III) to iron(II) during heat up, and (3) water formation during the thermolysis, which was shown to have a measurable impact on particle size and uniformity.To investigate further the particle development during this stage, several experiments were performed to evaluate pre-turbidity event particles and post-turbidity event particles. In the first experiment, TEM analysis of aliquots taken at 15, 30, 45, 60, 75 minutes at 324 °C, did not show any nanoparticles. A second experiment was performed to evaluate particle growth after nucleation, aliquots were taken 4 minutes before the turbidity event, at the turbidity event, turbidity + 4 min, turbidity + 8 min, turbidity + 30 min, and 24 h (post-oxidation) and characterized by TEM, DLS and VSM. From the TEM and DLS growth profiles, we observed nucleation occurred before the turbidity event, and particles grew rapidly for about 10 minutes after the turbidity event, reaching 95% of their final size, at which point the growth slowed. Bright field TEM images of selected samples from this series are provided in Fig. 6a–d, with corresponding selected area electron diffraction images shown in Fig. 5e and f. Interestingly, nanoparticles nucleated before the turbidity event and grew to 16 nm diameter by the first sample point, taken about 10 minutes after the temperature inflection that is presumed to coincide with nucleation. Particle size was 20 nm at the turbidity event (87% of final size), then growth began to slow 4 min after the turbidity event as the nanoparticles approached their final size. The TEM images (Fig. 5a–d) also show a notable shape evolution with time, after beginning with irregular shapes, reflecting rapid kinetically-driven growth, nanoparticles became rounder and more uniform during subsequent aging.
 | ||
| Fig. 5 Nanocrystal evolution during the thermolysis reaction. Bright field TEM images of sample aliquots (a–d), with selected area electron diffraction (e–h) Note that (d) and (h) represent nanoparticles after the oxidation step, which is discussed in Section 3.3. (i) Time evolution of particle size measured by TEM and DLS. DLS data is not shown for the final oxidized sample, since the strongly magnetic particles aggregated. (j) Magnetization curves measured at 295 K. | ||
 | ||
| Fig. 6 Bright field TEM images of nanoparticles with varying diameter (a) 15 nm, (b) 20 nm, (c) 24 nm, (d) 27 nm, (e) 30 nm, (f) 35 nm. In (g), size distributions are represented after fitting histograms to a log-normal distribution function. The legend lists the median diameter, d0 and the shape parameter, σ. | ||
As evidenced from the diffraction patterns (Fig. 5e–g), nanoparticles were mixed phase prior to oxidation, containing mostly wüstite during the early stages of nucleation and growth, but with spinel iron oxide also present, presumably as an oxidized shell. Nanoparticles displayed a spatially varying contrast in TEM, with a dark center and noticeable shell (Fig. 5a–c); in the earliest sample, wüstite was predominant (Fig. 5e), but later (30 min) a mix of wüstite and spinel iron oxide was evident (Fig. 5g). After oxidation, diffraction indicated pure magnetite (Fig. 5h). Magnetic measurements (Fig. 5j) also suggest the nanoparticles are mixed-phase before oxidation, with a weak superparamagnetic response that is attributed to an oxidized shell, but low moment per iron due to the antiferromagnetic wüstite core. The moment increased modestly with aging time, which may be because the oxidized shell volume increased with particle diameter. After oxidation, the magnetic properties indicate complete transformation to magnetite.
Of some importance is the finding that DLS analysis of the wüstite particles can accurately size the particles produced during the thermolysis. Excellent correlation between the DLS and TEM measurements was observed (Fig. 5i) (magnetic size measurements such as Chantrell fitting were not reliable for the mixed-phase pre-oxidized samples, since they assume a uniformly ferrimagnetic sample); the DLS size includes the oleic acid layer, which contributes about 2.5 nm to the measured diameter. DLS analysis of a pre-oxidation sample will allow the adjustment of the subsequent oxidation procedure to account for the size of the nanoparticles. The DLS measurement of oxidized magnetite particles was not successful due to apparent aggregation issues, presumably caused by their larger magnetic moment.
Finally, the reaction conditions (oleic acid ratio) could be varied to produce a range of particle sizes from 15 to 35 nm diameter with good uniformity. Fig. 6 shows TEM images of representative iron oxide nanoparticles within this range.
 | ||
| Fig. 7 Transformation of Fe(III) to Fe(II) during the heat up stage of thermolysis. | ||
 | ||
| Fig. 8 Proposed formation of undecenes following oxidative decarboxylation of iron oleate. | ||
Detection of these undecenes strongly suggests oxidative decarboxylation is operating during thermolysis. Since there is a very large amount of iron in solution, many of these processes likely occur through alkyl iron intermediates and iron–alkene coordinated complexes.
In the first experiment, side-by-side nanoparticle synthesis reactions (6.7:1 oleic acid to Fe) were performed using 1-octadecene in one reaction and octadecane in the other reaction. Surprisingly, a dramatic influence on size was observed: the 1-octadecene reaction produced 24.7 nm median diameter particles and the octadecane reaction produced 14.4 nm particles. This result demonstrates 1-octadecene, and/or its degradation products are directly involved in the mechanism of nanoparticle formation. As a benefit of the discovery that octadecane decreases the size of the particles, we performed a thermolysis reaction with an oleic acid to Fe ratio of 2:1 and obtained high quality 4.2 nm nanoparticles.
To determine the influence of 1-octadecene on the rate of Fe(II) formation during the heat-up stage of the thermolysis, a 10 mmol iron oleate reaction with octadecane was heated to 320 °C and then cooled under argon. During the heat-up stage of the thermolysis reaction both reactions lightened in color. At 315 °C, the 1-octadecene reaction was the typical clear tea color, but the octadecane reaction was slightly darker, cola in color. Potassium permanganate titration of this test reaction indicated 96% Fe(II) for octadecane vs. 102% Fe(II) when 1-octadecene was the solvent. On the basis of this result we conclude the majority of the reduction of Fe(III) during the heat up stage of the reaction is being performed via oxidative decarboxylation of oleic acid.
The first issue was a practical problem where water liberated from the heated reaction mixture condensed in the upper portion of the reaction vessel. The water would then drip into the reaction mixture, which, at temperatures greater than 250 °C, caused a vigorous and dangerous vaporization. This situation not only introduced randomness into the nanoparticle formation, something we needed to avoid, but it also resulted in severe pressure changes, resulting in air being sucked back into the reaction or the vessel being blown apart at the joints. To reduce the formation of water condensate, a stream of argon was introduced into the reaction vessel to help move the water vapor out of the flask and safely into a bump trap. After the reaction had reached 318 °C the argon stream was discontinued as the water was then removed during reflux by 1-octadecene vapor. While the reaction was heating up and during the time before nucleation and nanoparticle growth occurs, water evolved from the reaction mixture and was observed as a white cloud above the surface of the heated reaction. The white cloud deposited a mixture of 1-octadecene (5–10 mL) and water (∼3.2 mL, ∼180 mmol) into the bump trap. The amount of water vapor increased with the amount of oleic acid used in the reaction.
Much of the water observed during heat up was formed from oleic acid via a ketonization reaction to produce oleone (9(Z),26(Z)-pentatriacont-9,26-dien-18-one), water and carbon dioxide. Ketonization is known to occur to calcium oleate at 300 °C (for a leading ketonization reference see ref. 44). Davis and Schultz reported the decomposition of iron(II) carboxylates at 250–300 °C to produce symmetrical ketones, including iron(II) decanoate to produce 10-nonadecanone in 96% yield.45 They also observed that iron(III) carboxylates did not form ketones, from which we can conclude that some water is released only during heat up, after onset of iron(III) reduction discussed previously. To verify ketonization was occurring during the thermolysis of iron oleate, a reaction (40 mmol Fe, 7.2:1 oleic acid to Fe) was halted one hour after nucleation had occurred and a portion of this reaction mixture (1/37th total volume, 10 mL) was treated to flash chromatography. The ketone was isolated and identified by 1H-NMR (see supplemental material), demonstrating the reaction had produced 166 mmol of ketone (formed from 332 mmol of oleic acid). The starting reaction mixture contained 288 mmol of oleic acid and 120 mmol of oleate from iron oleate; therefore, the ketonization pathway accounts for at least 82% of the oleic acid decomposition. Of critical importance is the co-generation of 166 mmol of water during the ketonization of oleic acid. This generated water is likely first formed as iron oxides or iron hydroxides, which ligand exchange with oleic acid to liberate free water. It is currently believed the water produced by the ketonization of oleic acid plays a major role during nucleation and in the resulting nanoparticle size. As a consequence of oleic acid ketonization the acidity of the reaction mixture changes during the reaction. Under the initial acidic conditions the formation of iron oxides is disfavored by the acid. As the oleic acid transforms to ketone the reduced concentration of acid begins to allow formation of iron oxides. This drop in oleic acid concentration to a critical level is believed to initiate nucleation and growth of iron oxide nanoparticles.
Previous studies have shown water included during thermolysis influences nanoparticle size,46 and we also noticed controlling water content in the thermolysis improved size control and reproducibility. In order to institute better controls over the nanoparticle synthesis, we implemented inert atmosphere procedures to limit the amount of oxygen in the reaction mixture using a standard vacuum and purge protocol. It was noticed that during the evacuation a large amount of foaming in the reaction mixture occurred, presumably from acid induced ligand exchange and vaporization of released water. Holding the reaction under vacuum at 40 °C until all signs of gas evolution ceased, about 25 minutes, resulted in a noticeable improvement in size dispersity and reproducibility (Fig. 9). It was also accompanied by a shift in nanoparticle size, requiring the use of a higher oleic acid to iron ratio to obtain particles with diameter of about 25 nm. We realized such a large shift in particle size based on a 25 minute vacuum period could easily introduce reproducibility issues in size control. We therefore increased the time under vacuum to produce a more consistent reaction mixture for the thermolysis reaction. The use of vacuum for 18 hours at 50 °C, which removed about 250 mg of mass (presumably water, about 14 mmol), resulted in an additional improvement in size reproducibility of the nanoparticles formed in the thermolysis reaction. In Fig. 9, plots of measured nanoparticle diameter (TEM) vs. oleic acid ratio in the reaction mixture are provided with grouping by three different reaction pre-treatments. Linear fits indicate their relative reproducibility, where the R2 value of the reactions evacuated for 18 hours is twice that of the reactions with short or no evacuation. Other groups have also developed pre-treatments for the thermolysis of iron oleate.46,47
 | ||
| Fig. 9 Nanoparticle size and size distribution varied with water content in the reaction. | ||
A further shift in nanoparticle size was observed between the set of samples treated with 30 minutes of evacuation time and those that received 18 hours of evacuation time. Compared to no evacuation, an additional 44 mmol of oleic acid was required to achieve the same nanoparticle diameter, using the best fit lines in Fig. 9 (based on a ratio of 6.3 for no evacuation and 7.4 for 18 hours evacuation). This corresponds to 22 mmol of water generated by ketone formation, which compares to the 14 mmol removed during evacuation, accounting for some additional water loss during heat up of the reaction.
3.3. Oxidation to Fe3O4 with 1% oxygen in argon
Thermolysis of iron oleate in oleic acid under an inert atmosphere favors formation of antiferromagnetic wüstite nanoparticles, as observed in the sampled time-points presented in Fig. 5. A “clean” batch of wüstite particles, with minimal oxygen exposure for example from inserting a needle for sampling, was prepared by stopping a 40 mmol Fe reaction after 2.5 hours at 324 °C and cooling under argon. High quality wüstite particles (27.6 nm (0.08)) were obtained (see Fig. 10). Potassium permanganate titration showed these wüstite nanoparticles were 95% Fe(II). As seen in the bright field TEM image and the selected area electron diffraction in Fig. 10, the particles have a wüstite core and a thin inverse spinel shell. The inverse spinel rings are notably more diffuse, in contrast to the sharp wüstite rings, suggesting small magnetite crystallites due to oxidation of a thin shell on the particle surface. It is believed the magnetite shell forms during sample preparation. | ||
| Fig. 10 Wüstite nanoparticles obtained by thermolysis under argon for 2.5 hours at 324 °C without an oxidation step. (a) Bright field, in which a core/shell structure is visible, (b) SAED pattern, and (c) radial integration of the diffraction. Indexed rings correspond to wüstite unless noted. The magnetite rings are notably more diffuse, in contrast to the sharp wüstite rings, suggesting small magnetite crystallites due to oxidation of a thin shell on the particle surface. | ||
Interestingly, wustite formation can go unnoticed during small-scale thermolysis common to many previous studies, (e.g. 2.5 mmol Fe, 100 mL reactor), since the small quantity of oxygen needed to transform the wüstite particles to inverse-spinel iron oxide can be supplied via minor leaks in the apparatus, such as dry ground-glass joints. However, a dedicated oxidation step is needed to achieve this transformation at larger scale (e.g. 40 mmol) and prepare magnetite nanoparticles with optimized magnetic moment. Other oxidants reported in the literature such as 3-chloroperbenzoic acid and trimethylamine N-oxide were considered too toxic and/or hazardous. Other groups have used oxygen9 or air oxidation;48 however, introducing air to the thermolysis at 318 °C would be dangerous at large scale since 1-octadecene auto-ignites at ∼250 °C. Cooling the reaction for air-oxidation could be safer, and we determined air oxidation of wüstite nanoparticles to magnetite nanoparticles proceeded at 175 °C and was rapid at 200 °C, with the resulting particles having a clear electron diffraction pattern of magnetite. However, though below the auto-ignition temperature of octadecene, the addition of air to octadecene was still above the limiting oxygen concentration (LOC), making fire or explosions a concern for scalability.
To address these concerns, we developed an oxidation procedure using 1% oxygen in argon that is effective, simple, scalable, and can be performed safely at 318 °C by maintaining oxygen well below the estimated LOC.49 Following this procedure, after wüstite nanoparticle formation had occurred, 1% oxygen in argon was bubbled into the thermolysis reaction mixture while maintaining the reaction temperature of 318 °C. After optimizing the oxidation conditions for particle size, the resulting particles typically were highly magnetic, with saturation magnetization (Ms) of up to 80 emu g−1 (412 kA m−1), or 92% of bulk magnetite. Further analysis of the nanoparticles by X-ray and electron diffraction indicated they were composed of high purity magnetite. Typically, 1% oxygen was first added at ∼140 mL min−1 for 3 hours to perform most of the oxidation. To ensure the oxidation was complete without over-oxidizing the particles to maghemite, the flow of 1% oxygen was then reduced (15 mL min−1) for an additional 28 hours (at 318 °C).
As anticipated, the size of the nanoparticles affected the oxidation rate. On the scale of 40 mmol Fe, only 6.7 mmol is theoretically required to transform wüstite to magnetite. In practice, small (20.5 nm) particles required about 24 mmol of oxygen to obtain pure magnetite, whereas larger particles (26.9 nm) required about 64 mmol of oxygen. The excess oxygen is either not absorbed or is consumed oxidizing the 1-octadecene.
In Section 3.2, we presented results indicating 1-octadecene plays a small role in the reduction rate of iron(III) to iron(II) during thermolysis. Unsurprisingly, 1-octadecene is also oxidized along with nanoparticles during the oxidation step. We demonstrated the solvent influence by dispersing nanoparticles in 1-octadecene and octadecane and exposing both to air at 175 °C for 30 minutes. The 1-octadecene reaction remained black; however the octadecane reaction turned orange-red in color and lost all traces of black. In addition to color variations, we characterized the particles with electron diffraction. While the diffraction patterns of maghemite and magnetite feature substantial overlap, some fine features can be resolved if the crystal size is sufficiently large.53,54 Selected area electron diffraction of these samples (Fig. 11) show slight variations, including the (210) and (213) maghemite rings, which are visible only in the octadecane-oxidized sample. This result suggests 1-octadecene is also being oxidized during the oxidation step, preventing conversion of magnetite to maghemite. This effect could serve as an important oxidative buffer, preventing over oxidation of iron oxide nanoparticles.
 | ||
| Fig. 11 Selected area electron diffraction of nanoparticles oxidized in octadecene (a) and octadecane (b), and (c) radial integrations of the diffraction patterns (integrating 40° centered on 270°). Particles oxidized in octadecane (b) feature unique diffraction rings at 2.7 and 2.9 nm−1, which match reference patterns for maghemite. | ||
4. Conclusions
We have described a thermolysis procedure for making monodisperse magnetite nanoparticles with diameter between 15 and 35 nm, starting with iron(III) oleate as iron precursor and 1-octadecene as solvent and incorporating an in situ oxidation step for magnetic optimization. Investigations showed the iron oleate precursor consists of a triironoxonium structure. Reduction of iron(III) to iron(II) was shown to occur during the heat up phase of the reaction and proceeded via an oxidative decarboxylation pathway. The role of water in the starting iron oleate and generated during the ketonization was shown to influence the particle size and reproducibility. In addition to controlling nanoparticle size by addition of excess oleic acid, we introduce an in situ oxidation step with 1% O2 in argon to control the iron oxide phase and produce high purity crystalline magnetite. The use of 1% O2 in argon can safely translate to large scale, since it maintains oxygen concentration less than the LOC. The evaluation of iron(II) content using a permanganate titration procedure, optimized for oleic acid coated iron oxide nanoparticles, was performed. Thorough testing of this magnetite nanoparticle synthesis procedure indicated it is reproducible and amenable to large scale.Acknowledgements
We gratefully acknowledge funding from NIH grants R41EB013520 and R42EB013520 and NSF grant IIP 1215556.References
- Y. Bao, T. Wen, A. C. S. Samia, A. Khandhar and K. M. Krishnan, J. Mater. Sci., 2015, 1–41 Search PubMed.
- K. M. Krishnan, IEEE Trans. Magn., 2010, 46, 2523–2558 CrossRef CAS PubMed.
- A. P. Khandhar, R. M. Ferguson, J. A. Simon and K. M. Krishnan, J. Biomed. Mater. Res., Part A, 2012, 100, 728–737 CrossRef PubMed.
- L. M. Bauer, S. F. Situ, M. A. Griswold and A. C. S. Samia, J. Phys. Chem. Lett., 2015, 2509–2517 CrossRef CAS PubMed.
- R. M. Ferguson, A. P. Khandhar, S. J. Kemp, H. Arami, E. U. Saritas, L. R. Croft, J. Konkle, P. W. Goodwill, A. Halkola, J. Rahmer, J. Borgert, S. M. Conolly and K. M. Krishnan, IEEE Trans. Med. Imag., 2015, 34, 1077–1084 CrossRef PubMed.
- A. Demortière, P. Panissod, B. P. Pichon, G. Pourroy, D. Guillon, B. Donnio and S. Bégin-Colin, Nanoscale, 2011, 3, 225–232 RSC.
- S. A. Shah, D. B. Reeves, R. M. Ferguson, J. B. Weaver and K. M. Krishnan, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 92, 094438 CrossRef PubMed.
- T. Hyeon, S. S. Lee, J. Park, Y. Chung and H. B. Na, J. Am. Chem. Soc., 2001, 123, 12798–12801 CrossRef CAS PubMed.
- S. Sun and H. Zeng, J. Am. Chem. Soc., 2002, 124, 8204–8205 CrossRef CAS PubMed.
- S. Sun, H. Zeng, D. B Robinson, S. Raoux, P. M. M Rice, A. Shan, X. Wang and G. Li, 2004, 126, 273–279.
- W. Yu, J. Falkner, C. Yavuz and V. Colvin, Chem. Commun., 2004, 2306–2307 RSC.
- N. R. Jana, Y. Chen and X. Peng, Chem. Mater., 2004, 16, 3931–3935 CrossRef CAS.
- J. Park, K. An, Y. Hwang, J.-G. Park, H.-J. Noh, J.-Y. Kim, J.-H. Park, N.-M. Hwang and T. Hyeon, Nat. Mater., 2004, 3, 891–895 CrossRef CAS PubMed.
- E. C. Vreeland, J. Watt, G. B. Schober, B. G. Hance, M. J. Austin, A. D. Price, B. D. Fellows, T. C. Monson, N. S. Hudak, L. Maldonado-Camargo, A. C. Bohorquez, C. Rinaldi and D. L. Huber, Chem. Mater., 2015, 27, 6059–6066 CrossRef CAS.
- G. A. Bain and J. F. Berry, J. Chem. Educ., 2008, 85(4), 532–536 CrossRef CAS.
- P. Pascal, Ann. Chim. Phys., 1910, 19, 5–70 CAS.
- R. Hufschmid, H. Arami, R. M. Ferguson, M. Gonzales, E. Teeman, L. N. Brush, N. D. Browning and K. M. Krishnan, Nanoscale, 2015, 7, 11142–11154 RSC.
- F. Ozel, H. Kockar, S. Beyaz, O. Karaagac and T. Tanrisever, J. Mater. Sci.: Mater. Electron., 2013, 24, 3073–3080 CrossRef CAS.
- L. M. Bronstein, X. Huang, J. Retrum, A. Schmucker, M. Pink, B. D. Stein and B. Dragnea, Chem. Mater., 2007, 19, 3624–3632 CrossRef CAS.
- S. Palchoudhury, W. An, Y. Xu, Y. Qin, Z. Zhang, N. Chopra, R. A. Holler, C. H. Turner and Y. Bao, Nano Lett., 2011, 11, 1141–1146 CrossRef CAS PubMed.
- S. M. Oh, D. N. Henderickson, K. L. Hassett and R. E. Davis, J. Am. Chem. Soc., 1985, 107, 8009–8018 CrossRef CAS.
- H. B. Abrahamson and H. C. Lukaski, J. Inorg. Biochem., 1994, 54, 115–130 CrossRef CAS.
- A. B. Blake and L. R. Fraser, J. Chem. Soc., Dalton Trans., 1975, 193 RSC.
- T. Nakamoto, M. Katada, K. Endo and H. Sano, Polyhedron, 1998, 17(20), 3507–3514 CrossRef CAS.
- C.-J. Chen, H.-Y. Lai, C.-C. Lin, J.-S. Wang and R.-K. Chiang, Nano Res. Lett., 2009, 4, 1343–1350 CrossRef CAS PubMed.
- A. Earnshaw, B. N. Figgis and J. Lewis, J. Chem. Soc. A, 1966, 1656 RSC.
- G. Novitchi, L. Helm, C. Anson, A. K. Powell and A. E. Merbach, Inorg. Chem., 2011, 50, 10402–10416 CrossRef CAS PubMed.
- M. R. Silva, J. N. J. Nogueira, P. A. O. C. Silva, C. Yuste-Vivas, L. C. J. Pereira and J. C. Waerenborgh, Solid State Phenom., 2012, 194, 162–170 CrossRef.
- A. K. Boudalis, Y. Sanakis, C. P. Raptopoulou, A. Terzis, J.-P. Tuchagues and S. P. Perlepes, Polyhedron, 2005, 24, 1540–1548 CrossRef CAS.
- H. T. Hai, H. T. Yang, H. Kura, D. Hasegawa, Y. Ogata, M. Takahashi and T. Ogawa, J. Colloid Interface Sci., 2010, 346, 37–42 CrossRef CAS PubMed.
- B. P. Pichon, O. Gerber, C. Lefevre, I. Florea, S. Fleutot, W. Baaziz, M. Pauly, M. Ohlmann, C. Ulhaq, O. Ersen, V. Pierron-Bohnes, P. Panissod, M. Drillon and S. Begin-Colin, Chem. Mater., 2011, 23, 2886–2900 CrossRef CAS.
- Y. Hou, Z. Xu and S. Sun, Angew. Chem., 2007, 46, 6329–6332 CrossRef CAS PubMed.
- F. X. Redl, C. T. Black, G. C. Papaefthymiou, R. L. Sandstrom, M. Yin, H. Zeng, C. B. Murray and S. P. O'Brien, J. Am. Chem. Soc., 2004, 126, 14583–14599 CrossRef CAS PubMed.
- C.-J. Chen, R.-K. Chiang, H.-Y. Lai and C.-R. Lin, J. Phys. Chem. C, 2010, 114, 4258–4263 CAS.
- R. Chen, M. G. Christiansen, A. Sourakov, A. Mohr, Y. Matsumoto, S. Okada, A. Jasanoff and P. Anikeeva, Nano Lett., 2016, 16(2), 1345–1351 CrossRef CAS PubMed.
- S. G. Kwon, Y. Piao, J. Park, S. Angappane, Y. Jo, N.-M. Hwang, J. G. Park and T. Hyeon, J. Am. Chem. Soc., 2007, 129, 12571–12584 CrossRef CAS PubMed.
- F. van der Klis, M. H. van den Hoorn, R. Blaauw, J. van Haveren and D. S. van Es, Eur. J. Lipid Sci. Technol., 2011, 113, 562–571 CrossRef CAS.
- T. Ueno, E. Nakashima and K. Takeda, Polym. Degrad. Stab., 2010, 95, 1862–1869 CrossRef CAS.
- M. R. Bukowski, P. Comba, A. Lienke, C. Limberg, C. Lopez de Laorden, R. Mas-Ballesté, M. Merz and L. Que, Angew. Chem., Int. Ed. Engl., 2006, 45, 3446–3449 CrossRef CAS PubMed.
- K. Hasan, N. Brown and C. M. Kozak, Green Chem., 2011, 13, 1230–1237 RSC.
- K. Chen and L. Que Jr, Angew. Chem., Int. Ed., 1999, 38, 2227–2229 CrossRef CAS.
- T. W.-S. Chow, E. L.-M. Wong, Z. Guo, Y. Liu, J.-S. Huang and C.-M. Che, J. Am. Chem. Soc., 2010, 132, 13229–13239 CrossRef CAS PubMed.
- E. Ertürk, M. A. Tezeren, T. Tilki and T. Erdogan, Polymer, 2012, 66, 2373–2377 Search PubMed.
- T. N. Pham, T. Sooknoi, S. P. Crossley and D. E. Resasco, ACS Catal., 2013, 3, 2456–2473 CrossRef CAS.
- R. Davis and H. P. Schultz, J. Org. Chem., 1962, 27, 854–857 CrossRef CAS.
- L. M. Bronstein, J. E. Atkinson, A. G. Malyutin, F. Kidwai, B. D. Stein, D. G. Morgan, J. M. Perry and J. A. Karty, Langmuir, 2011, 27, 3044–3050 CrossRef CAS PubMed.
- W. Baaziz, B. P. Pichon, S. Fleutot, Y. Liu, C. Lefevre, J.-M. Greneche, M. Toumi, T. Mhiri and S. Begin-Colin, J. Phys. Chem. C, 2014, 118, 3795–3810 CAS.
- K. Woo, J. Hong, S. Choi, H.-W. Lee, J.-P. Ahn, A. Chul Sung Kim and S. W. Lee, Chem. Mater., 2004, 16, 2814–2818 CrossRef CAS.
- P. M. Osterberg, J. K. Niemeier, C. J. Welch, J. M. Hawkins, J. R. Martinelli, T. E. Johnson, T. W. Root and S. S. Stahl, Org. Process Res. Dev., 2014, 141223071236001 Search PubMed.
- L. Finkelstein, The titration of iron by potassium permanganate, Armour Institute of Technology, 1914 Search PubMed.
- J. Santoyo Salazar, L. Perez, O. de Abril, L. Truong Phuoc, D. Ihiawakrim, M. Vazquez, J.-M. Greneche, S. Begin-Colin and G. Pourroy, Chem. Mater., 2011, 23, 1379–1386 CrossRef CAS.
- R. Frison, G. Cernuto, A. Cervellino, O. Zaharko, G. M. Colonna, A. Guagliardi and N. Masciocchi, Chem. Mater., 2013, 25, 4820–4827 CrossRef CAS.
- K. Haneda and A. H. Morrish, Solid State Commun., 1977, 22, 779–782 CrossRef CAS.
- R. M. Cornell and U. Schwertmann, The Iron Oxides, John Wiley & Sons, 2003 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Discussion of iron oleate synthesis parameters and impact on nanoparticle morphology, discussion of TLC analysis of thermolysis reaction mixture, discussion of nanoparticle size reproducibility and stirring rate, powder X-ray diffraction of sample NP5, 1H NMR of mixed fatty ketones extracted from bump trap during thermolysis reaction. See DOI: 10.1039/c6ra12072e |
| This journal is © The Royal Society of Chemistry 2016 |