
Tetranuclear zinc cluster: a dual purpose catalyst for per-O-acetylation and de-O-acetylation of carbohydrates†‡
Ting-Wei Lin§
,
Avijit K. Adak§,
Hong-Jyune Lin,
Anindya Das,
Wei-Chen Hsiao,
Ting-Chun Kuan and
Chun-Cheng Lin*
Department of Chemistry, National Tsing Hua University, Hsinchu-300, Taiwan. E-mail: cclin66@mx.nthu.edu.tw; Fax: +886 3 5711082; Tel: +886 3 5753147
First published on 6th June 2016
Abstract
The trifluoroacetic acid adduct of tetranuclear zinc cluster Zn4(OCOCF3)6O catalysis in per-O-acetylation and de-O-acetylation of carbohydrates at 70 °C can be tuned by adjusting the reaction medium. Per-O-acetylation of hexopyranoses with a near stoichiometric amount of acetic anhydride in toluene resulted in the exclusive formation of pyranosyl products as an anomeric mixture, whereas de-O-acetylation of acetates occurred in methanol in high yields. In the latter, methanol acts as both nucleophile and solvent, and the reaction conditions were compatible to acid- and base-sensitive groups and amino acid derivatives.
Introduction
Hydroxyl functionality is ubiquitous in natural products and, consequently, requires the introduction of O-acetate as a mask for hydroxyl groups and subsequent deprotection steps.1 In the context of carbohydrate chemistry, O-acetyl groups not only serve as protecting groups, but also as modulators of reactivity in glycosylation. Per-O-acetylated carbohydrates are inexpensive and invaluable substrates for the preparation of various biologically relevant oligosaccharides, glycoconjugates, and natural products.2 In addition, per-O-acetylation is routinely employed for characterization and identification of carbohydrate structures. As a consequence, an overwhelming number of O-acetylation and de-O-acetylation methods have been developed for widespread use in organic and natural product synthesis, as well as industrial processes.3 The standard O-acetylation reaction is typically carried out using either acetyl chloride or acetic anhydride as the acetylating reagent. The latter is perhaps the most extensively used reagent, and usually requires a catalyst to achieve a reasonable reaction rate. For per-O-acetylation of carbohydrates, pyridine is the most commonly used solvent/catalyst, although it is known to have unpleasant odors and acute toxicity.4 Certain derivatives of pyridine such as 4-(N,N-dimethylamino)pyridine (DMAP) and 4-(1-pyrrolidino)pyridine also catalyze the acetylation reaction, in some instances 104-times faster than pyridine.5 Sodium acetate6 and iodine7 are also used as catalysts in the per-O-acetylation of carbohydrates. Other reagents that catalyze O-acetylation of carbohydrates using acetic anhydride include Lewis acids, for instance Ce(OTf)2,8 ZnCl2,9 FeCl3,10 V(O) (OTf)2,11 Sc(OTf)3,12 Cu(OTf)2,13 Zn(ClO4)2·6H2O,14 Cu(ClO4)2·6H2O;15 Brønsted acids such as HClO4,16 H2SO4,17 sulfamic acid,18 and methanesulfonic acid;19 heterogeneous catalysts such as montmorillonite K-10,20 H-beta zeolite,21 molecular sieves,22 ionic liquid,23 enzyme catalysts such as lipases,24 and others such as deep eutectic solvent,25 and sulfonic acid-functionalized nano γ-Al2O3.26 Although these methods are effective for per-O-acetylation of carbohydrates, the use of excess acetic anhydride as solvent causes tedious work up in the neutralization step. Additionally, only a few of these have been used in large-scale synthesis of carbohydrate building blocks and intermediates.10,13,19In this context, previously we introduced LiClO4 as a mild catalyst in combination with a stoichiometric amount of acetic anhydride for per-O-acetylation of saccharides under solvent-free conditions, scalable and applicable to multi-gram synthesis.27 For preparative purposes, later we carried out synthesis of per-O-acetylated saccharides on a multi-gram scale using catalytic LiClO4, involving only a simple work-up and neutralization with saturated aqueous NaHCO3 solution to give products.28
Here, we report the first use of a μ-oxo-tetranuclear zinc cluster trifluoroacetic acid adduct Zn4(OCOCF3)6O·(CF3COOH)n (1)29e as a dual catalyst for per-O-acetylation of saccharides using near-stoichiometric acetic anhydride in toluene, and de-O-acetylation of saccharides when methanol is used as a solvent and nucleophile. The Zn4(OCOCF3)6O catalyst has been employed in a number of synthetic applications, including the acylation of alcohols and deacylation of acetates and benzoates.29,30 To our knowledge, this is the first use of 1 for per-O-acetylation and de-O-acetylation of carbohydrates, considerably expanding the application scope of Zn cluster catalysis in carbohydrate chemistry.
In the seminal work of Ohshima and Mashima,29,30 tetranuclear Zn cluster was utilized as a robust multimetallic catalyst that enables acylation of alcohols in the presence of amines. The selective transesterification of hydroxyl groups in the presence of primary and secondary amines might explain the extremely high oxophilicity of this Zn catalysis. Despite this high chemoselectivity in O-acylation reaction of the amino alcohols and transesterification of amino acid ester derivatives, the only monosaccharide tested30b has largely been limited to acetylation of the C6 primary hydroxyl group. Therefore more studies are needed to understand the substrate scope of this catalysis. Furthermore, catalyst 1 might be susceptible to other classes of (bio)molecules using different acylation reagents such as acetic anhydride. To address this, we investigated enhanced enzyme-like selectivity of 1 for O-acetylation and de-O-acetylation of hydroxyl groups to structurally more diverse polyhydroxylated compounds such as carbohydrates and also amino acid derivatives.
We were pleased to see that exposure of methyl-α-D-glucopyranoside (2) to a near-stoichiometric amount of acetic anhydride (1.1 mol eq. per OH group of sugar) in the presence of tetranuclear Zn catalyst 1 (1.25 mol% per OH group of sugar) and at 70 °C for 12 h (indicated by the disappearance of 2 on TLC) did indeed provide the fully acetylated methyl-α-D-glucopyranoside (10)31 as essentially a single component, without any observed effect on the interglycosidic linkage and with good yield (Table 1, entry 1, 99% yield). The product could be isolated with minimal handling and purifications; removal of volatiles under reduced pressure was followed by passing through a short plug of silica to remove the catalyst with a purity of more than 97%, as indicated by 1H NMR spectroscopy. Further examination with a disaccharide, D-lactose (3), under identical conditions as described above also revealed the desired product in a yield of 98% (Table 1, entry 2). The excellent yields and efficiencies exhibited by the catalyst 1 prompted us to select these conditions for further exploration.
|
||||
|---|---|---|---|---|
| Entry | Substrate | Product | Crude yielda (%) | (α/β) |
a Yield of crude product with purity higher than 97% according to 1H NMR spectroscopy.b A mixture of D-galactopyranosyl pentaacetates and D-galactofuranosyl pentaacetates with the ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.c A mixture of D-mannopyranosyl pentaacetates and D-mannofuranosyl pentaacetates with the ratio 10:1. :1.c A mixture of D-mannopyranosyl pentaacetates and D-mannofuranosyl pentaacetates with the ratio 10:1. |
||||
| 1 |  |
 |
99 | — |
| 2 |  |
 |
98 | 0.7/1 |
| 3 |  |
 |
98 | 0.9/1 |
| 4 |  |
 |
97 | 1/2.2 |
| 5 |  |
 |
96 | — |
| 6 |  |
 |
98 | — |
| 7b |  |
 |
99 | 0.6/1 |
| 8c |  |
 |
99 | 1/0.9 |

Next, the scope for O-acetylation of hydroxyl groups in other saccharides was examined (Table 1). D-Glucose (4) and N-acetyl-D-glucosamine (5) provided a comparable yield of corresponding acetylated product in 98% (entry 3) and 97% (entry 4), respectively. Moreover, acid labile groups such as isopropylidene survived the present conditions (entry 5). As a representative example of a complex carbohydrate, N-acetylneuraminic acid methylester (7), a polyfunctional ulosonic acid of five hydroxyl groups with different reactivities, was acylated to 15 in which the hydroxyl at C-2 position remained unaffected (entry 6), in accord with previous reports.27 Compound 15 is a valuable precursor for the synthesis of sialyl phosphite donors,32 our new procedure should provide a desirable alternative. In Table 1, per-O-acetylated saccharides (entries 2–4) are exclusively in the pyranosyl form as an anomeric mixture, and the assignment of anomeric stereochemistry was determined based on 400 MHz 1H NMR spectral analyses. However, per-O-acetylation of hexoses with “galactosyl” and “mannosyl” configurations produced a mixture of pyranosyl forms together with undesired D-galactofuranosyl pentaacetates (pyranosyl/furanosyl = 1/1, entry 7) and D-mannofuranosyl pentaacetates (pyranosyl/furanosyl = 10/1, entry 8), as inspected by the 1H NMR spectra of the crude products.33 In short, we demonstrate the application of tetranuclear Zn catalyst 1 in per-O-acetylation of saccharides.
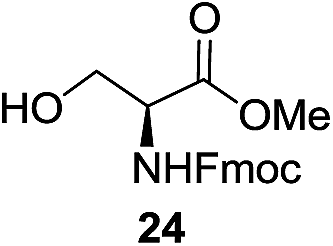
A diverse and representative scope of saccharides is well tolerated by this catalysis (Table 2) inclusive of anomeric azide (entry 1, 96%), and an 5N,4O-carbonyl-proteced thiosialoside (entry 3, 95%). However, in the case of partially benzylated lactoside (19) tethered with an alkyl azide at the reducing end, resulted in the formation of desired product (28) in a modest 40% yield (entry 2). The yield of 28 was diminished presumably due to the competing formation of a mixture of regioisomeric mono-O-acetylated derivative (32%). The addition of more than 1.25 mol% of 1 and a prolonged reaction time (30 h) did not alter the outcome of the reaction. Amino sugars which occur abundantly in natural oligosaccharides with varied substitutions at C-2 and anomeric position are successful in acylation reaction (entries 4–6). Extension of this protocol to N-trichloroethoxycarbonyl (Troc) protected thioglycosides 21–22 furnished fully protected thioglycosides 30 (entry 4) and 31 (entry 5) in excellent yields (92–97%). Moreover, a serine-derived glucoside (23) produced fully-protected serine glucoside 32 also in a respectable yield of 94% (entry 6), demonstrating the potential of Zn catalysis in glycoprotein synthesis. Finally, to examine the mildness of the acylation conditions, we also conducted O-acetylation of L-serine derivatives 24–25 (N-Fmoc) and 26 (N-Boc). As before, O-acetylation again accomplished excellent yields (33: 98%, 34: 93%, and 35: 95%; entries 7–9), and more importantly, neither epimerization of the α-stereocenter nor O-alkyl transesterification of carbamates was noticed. In addition, the catalytic system is susceptible to acid-sensitive groups such as benzyl and allyl ester, and Boc protecting group.



















Conversely, because of its efficiency and irreversible nature, de-O-acetylation of acetates in carbohydrates still relies heavily on classical basic or acidic hydrolysis, such as those that involve NaOMe (Zemplén deacylation),1 K2CO3,34 N2H4,35 DBU,36 and Brønsted acids like HCl.37 Recently, oxometallic species was employed at elevated temperature to facilitate deacylation of monosaccharides.38 Aqueous or pure alcohol is the preferred solvent in such de-O-acetylation reactions, and almost pure products could be obtained by a careful neutralization of the catalysts, usually by exposing to acid/base or acidic/basic resins followed by evaporation of the volatiles. These reaction protocols are useful; however, exposure to acid or base may have detrimental effects on substrates such as elimination and epimerization, and functional groups sensitive to acid or base may not be fully compatible. Therefore, a milder and neutral catalytic de-O-acetylation method which no longer requires such neutralization steps is highly desirable. In view of the transesterification of acetates using 1, we hypothesized that methanolysis of saccharides catalyzed by Zn cluster 1 could be performed, as 1 retained its catalytic activity even in the presence of excess amounts of alcohol.30
An initial evaluation of the proposed catalytic de-O-acetylation was conducted with 27 in the presence of 1.25 mol% (per OAc group of sugar) of 1, and transesterification was completed within 12 h (indicated by the disappearance of 27 on TLC) under refluxing methanol (0.6 M) (Table 3, entry 1). More importantly, because methylacetate is the only co-product formed in this process, pure (by 1H NMR) compound 18 could be obtained in a 91% isolated yield (entry 1) simply by evaporation and filtration through a short-pad of silica gel. De-O-acetylation of other important substrates under these conditions was then attempted and the results are shown in Table 3. The developed de-O-acetylation protocol can be conducted very easily and safely; the transesterification proceeded simply by heating the acetates in commercial methanol in the presence of a catalytic amount of 1. Thioglycosides (entries 2–5) with an N-Troc protection at C-2 (entries 2–3), and an N-benzyl-2,3-trans oxazolidinone (entry 4) are capable of deacylation by this catalytic protocol with consistently very good to excellent yields of products (89–98%). Implementation of a selenophenyl galactoside 38 with an azido group at C-2 (entry 6) proved sluggish (20 h) and needs 5 mol% catalyst to give a reasonably good yield of 87%. Protected serine–glucoside 39 can be similarly unmasked to triol 23 with the transesterification of allyl ester in a yield of 74% (entry 7). Removal of O-acetates in L-serine derivatives 33–35 readily provided corresponding products almost quantitatively (entries 8–10). While deacetylation of L-serine derivatives caused a transesterification of the benzyl (entry 8) and allyl ester (entries 9–10), respectively, neither epimerization of α-stereocenter nor O-alkyl transesterification of carbamates was observed.




















We next examined the substrate generality of the Zn cluster-catalyzed de-O-acetylation under refluxed methanol conditions (Table 4). The reaction protocol is straightforward and is susceptible to de-O-acetylation of a variety of saccharides. As revealed in Table 4, selectively protected hexo-furanose (entries 1 and 4), galacto- (entries 2 and 7), gluco- (entries 3, 5, and 6) and manno- (entry 8) pyranose derivatives can be efficiently unmasked in good to nearly quantitative yields (95–99%). Importantly, TBDMS, phthalimido (Phth), and acid sensitive groups such as isopropylidene (entries 1–4), trityl (entry 6), and benzylidene (entries 7–8) are tolerated in these reaction conditions without loss in reaction efficiencies.

















Conclusions
In summary, we have demonstrated that tetranuclear Zn cluster Zn4(OCOCF3)6O·(CF3COOH)n is an extremely effective dual purpose catalyst for per-O-acetylation and de-O-acetylation of carbohydrates. This method required only a near stoichiometric amount of acetic anhydride (1.1 mol equiv. per OH group of sugar) for O-acetylation of saccharides. The de-O-acetylation process can be performed very easily and safely; simply by heating the acetates in commercial grade methanol in the presence of catalytic amount of 1 (1.25 mol% per OH group of sugar). Overall, employing the tetranuclear Zn cluster as catalyst minimizes reagent use, workup, and purifications. We anticipate that this method of protection/deprotection strategy to prepare commonly used building blocks would find applications in oligosaccharide synthesis.Experimental section
General methods
The catalyst, tetranuclear Zn cluster trifluoroacetic acid adduct Zn4(OCOCF3)6O·(CF3COOH)n (1) (Strem Chemicals Inc., USA., Cat. no. 30-4050) was obtained from commercial source and used as received. All reactions were performed in oven-dried glassware (120 °C) under a nitrogen atmosphere unless otherwise specified. All solvents were dried and distilled by standard techniques. 1H and 13C NMR spectra were recorded on a Bruker AV-400 spectrometer operating at 400 MHz for 1H and 100 MHz for 13C, respectively. Chemical shifts (δ) are reported in ppm and referenced to the solvent used (CDCl3, δ 7.24 and 77.23; CD3OD, δ 3.31 and 49.0), with coupling constants (J) reported in Hz. High-resolution mass spectra were recorded using electrospray ionization mode with a time-of-flight detector. Thin-layer chromatography (TLC) analysis was monitored with 0.25 mm pre-coated plates (G60F254) and detected by UV absorption at 254 nm or by staining with p-anisaldehyde–sulfuric acid at 150 °C. Silica gel 60 (E. Merck) was employed for all flash-chromatography separations.General procedure for per-O-acetylation
A sealed tube was charged with sugar (0.51 mmol), acetic anhydride (1.1 eq. per OH), catalyst 1 (1.25 mol% per OH), and toluene (0.9 mL). The flask was heated at 70 °C for 12 h under an atmosphere of nitrogen. The volatiles were removed under reduced pressure, and the reaction mixture was passed through a short plug of silica gel to afford expected products in good to excellent yields (Tables 1 and 2).General procedure for de-O-acetylation
A mixture of 1 (1.25 mol% per OAc of sugar) and appropriate substrates (Tables 2 and 3) (0.51 mmol) in methanol (1.6 mL) was heated at reflux (70 °C, bath temperature) for 12 h under nitrogen. After removing volatiles under reduced pressure, the crude product was passed through a short plug of silica gel to provide desired products in moderate to excellent yields (Tables 3 and 4).Acknowledgements
We thank the National Tsing Hua University, Ministry of Education, and the Ministry of Science and Technology, Taiwan for financial support of this work.Notes and references
- (a) T. W. Greene and P. G. M. Wuts, Protective Groups in Organic Synthesis, Wiley, New York, 4th edn, 2006 Search PubMed; (b) P. J. Kocienski, Protecting Groups, Thieme, New York, 1994 Search PubMed.
- J. K. Baskin, Chem. Rev., 2000, 100, 4265–4266 CrossRef.
- (a) R. W. Dugger, J. A. Ragan and D. H. B. Ripin, Org. Process Res. Dev., 2005, 9, 253–258 CrossRef CAS; (b) J. S. Carey, D. Laffan, C. Thomsom and M. T. Williams, Org. Biomol. Chem., 2006, 4, 2337–2347 RSC.
- B. Yu, J. Xie, S. Deng and Y. J. Hui, J. Am. Chem. Soc., 1999, 121, 12196–12197 CrossRef CAS.
- G. Höfle, W. Steglich and H. Vorbrüggen, Angew. Chem., Int. Ed., 1978, 17, 569–583 CrossRef.
- J. Gelas, Adv. Carbohydr. Chem. Biochem., 1981, 39, 71–102 CrossRef CAS.
- K. P. R. Kartha and R. A. Field, Tetrahedron, 1997, 53, 11753–11766 CrossRef CAS.
- G. Bartoli, R. Dalpozzo, A. Nino, L. Maiuolo, M. Nardi, A. Procopio and A. Tagarelli, Green Chem., 2004, 6, 191–192 RSC.
- A. I. Vogel, Vogel's Textbook of Practical Organic Chemistry, Wiley, New York, 5th edn, 1989, pp. 644–651 Search PubMed.
- F. Dasguta, P. P. Singh and H. C. Srivastava, Carbohydr. Res., 1980, 80, 346–349 CrossRef.
- C.-T. Chen, J.-H. Kuo, C.-H. Li, N. B. Barhate, S.-W. Hon, T.-W. Li, S.-D. Chao, C.-C. Liu, Y.-C. Li, I.-H. Chang, J.-S. Lin, C.-J. Liu and Y.-C. Chou, Org. Lett., 2001, 3, 3729–3732 CrossRef CAS PubMed.
- M. Ishihara, M. Kubota, H. Kurihara and H. Yamamoto, J. Am. Chem. Soc., 1995, 117, 4413–4414 CrossRef.
- C.-A. Tai, S. S. Kulkarni and S.-C. Hung, J. Org. Chem., 2003, 68, 8719–8722 CrossRef CAS PubMed.
- G. Bartoli, M. Bosco, R. Dalpozzo, E. Marcantoni, M. Massaccessi and L. Sambri, Eur. J. Org. Chem., 2003, 4611–4617 CrossRef CAS.
- D. Chatterjee, A. Paul and S. Yadav, RSC Adv., 2015, 5, 29669–29674 RSC.
- A. K. Chakraborti and R. Gulhane, Chem. Commun., 2003, 1896–1897 RSC.
- J. A. Hyatt and G. W. Tindall, Heterocycles, 1993, 35, 227–234 CrossRef CAS.
- A. Santra, G. Guchhait and A. K. Mishra, Green Chem., 2011, 13, 1345–1351 RSC.
- S. K. Giri and K. P. R. Kartha, RSC Adv., 2015, 5, 11687–11696 RSC.
- P. M. Bhaskar and D. Loganathan, Tetrahedron Lett., 1998, 39, 2215–2218 CrossRef CAS.
- P. M. Bhaskar and D. Loganathan, Synlett, 1999, 129–131 CrossRef CAS.
- L. Cai, C. Rufty and M. Liquois, Asian J. Chem., 2014, 26, 4367–4369 CAS.
- S. A. Forsyth, D. R. MacFarlane, R. J. Thomson and von M. Itzstein, Chem. Commun., 2002, 714–715 RSC.
- N. Junot, J. C. Meslin and C. Rabiller, Tetrahedron: Asymmetry, 1995, 6, 1387–1390 CrossRef CAS.
- S. M. Rokade and P. M. Bhate, Carbohydr. Res., 2015, 416, 21–23 CrossRef CAS PubMed.
- L. Wu and Z. Yin, Carbohydr. Res., 2013, 365, 14–19 CrossRef CAS PubMed.
- K.-C. Lu, S.-Y. Hsieh, L. N. Patkar, C.-T. Chen and C.-C. Lin, Tetrahedron, 2004, 60, 8967–8973 CrossRef CAS.
- C.-C. Lin, L.-C. Huang, P.-H. Liang, C.-Y. Liu and C.-C. Lin, J. Carbohydr. Chem., 2006, 25, 303–313 CrossRef CAS.
- (a) T. Ohshima, T. Iwasaki, Y. Maegawa, A. Yoshiyama and K. Mashima, J. Am. Chem. Soc., 2008, 130, 2944–2945 CrossRef CAS PubMed; (b) T. Iwasaki, Y. Maegawa, Y. Hayashi, T. Ohshima and K. Mashima, J. Org. Chem., 2008, 73, 5147–5150 CrossRef CAS PubMed; (c) Y. Maegawa, K. Agura, Y. Hayashi, T. Ohshima and K. Mashima, Synlett, 2012, 23, 137–141 CAS; (d) Y. Maegawa, T. Ohshima, Y. Hayashi, K. Agura and K. Mashima, ACS Catal., 2011, 1, 1178–1182 CrossRef CAS; (e) Y. Hayashi, T. Ohshima, Y. Fujii, Y. Matsushima and K. Mashima, Catal. Sci. Technol., 2011, 1, 230–233 RSC.
- (a) T. Iwasaki, K. Agura, Y. Maegawa, Y. Hayashi, T. Ohshima and K. Mashima, Chem.–Eur. J., 2010, 16, 11567–11571 CrossRef CAS PubMed; (b) T. Iwasaki, Y. Maegawa, Y. Hayashi, T. Ohshima and K. Mashima, Synlett, 2009, 10, 1659–1663 Search PubMed.
- W. J. Goux and C. J. Unkefer, Carbohydr. Res., 1987, 159, 191–210 CrossRef CAS PubMed.
- C.-C. Lin, A. K. Adak, J.-C. Horng and C.-C. Lin, Tetrahedron, 2009, 65, 4714–4725 CrossRef CAS.
- V. Ferrières, M. Gelin, R. Boulch, L. Toupet and D. Plusquellec, Carbohydr. Res., 1998, 314, 79–83 CrossRef.
- J. J. Plattner, R. D. Gless and H. Rapoport, J. Am. Chem. Soc., 1972, 94, 8613–8615 CrossRef CAS PubMed.
- W. R. Roush and X.-F. Lin, J. Am. Chem. Soc., 1995, 117, 2236–2250 CrossRef CAS.
- L. H. B. Baptistella, J. F. Dos Santos, K. C. Ballabio and A. Marsaioli, Synthesis, 1989, 436–439 CrossRef CAS.
- (a) N. Yamamoto, T. Nishikawa and M. Isobe, Synlett, 1995, 505–506 CrossRef CAS; (b) V. Pozsgay, J. Am. Chem. Soc., 1995, 117, 6673–6681 CrossRef CAS.
- C.-Y. Liu, H.-L. Chen, C.-M. Ko and C.-T. Chen, Tetrahedron, 2011, 67, 872–876 CrossRef CAS.
Footnotes |
| † In memory of Dr Chang-Ching Lin. |
| ‡ Electronic supplementary information (ESI) available: Copies of NMR (1H and 13C) spectra of all new compounds. See DOI: 10.1039/c6ra12050d |
| § Authors have equal contribution. |
| This journal is © The Royal Society of Chemistry 2016 |