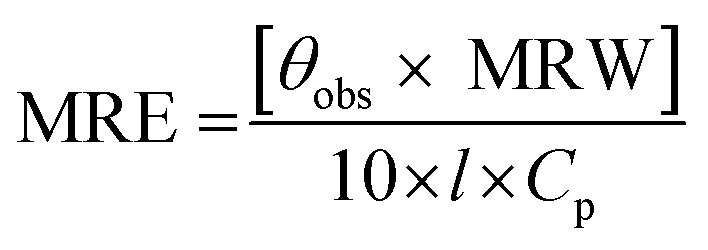DOI:
10.1039/C6RA12019A
(Paper)
RSC Adv., 2016,
6, 91756-91767
Interaction of an anticancer drug, gefitinib with human serum albumin: insights from fluorescence spectroscopy and computational modeling analysis†
Received
9th May 2016
, Accepted 19th September 2016
First published on 19th September 2016
Abstract
Binding of gefitinib (GEF), a promising anticancer drug to human serum albumin (HSA), the major transport protein in blood circulation was investigated using fluorescence, UV-vis absorption and circular dichroism (CD) spectroscopy as well as computational modeling. Fluorescence quenching of HSA upon GEF addition was found to be a static quenching process, as revealed from the decreasing trend of the Stern–Volmer quenching constant with increasing temperature as well as UV-vis absorption spectral results. Fluorescence quenching titration results demonstrated moderate binding affinity with the binding constant, Ka value as 1.70 × 104 M−1 between GEF and HSA at 15 °C. Thermodynamic data (ΔH = −7.74 kJ mol−1 and ΔS = +54.06 J mol−1 K−1) suggested participation of both hydrophobic interactions and hydrogen bonds in stabilizing the GEF–HSA complex, which was further supported by computational modeling results. The far-UV and the near-UV CD spectra showed secondary and tertiary structural changes in HSA, whereas three-dimensional fluorescence spectral results indicated microenvironmental perturbations around protein fluorophores upon GEF binding. Binding of GEF to HSA offered significant protection to the protein against thermal destabilization. Competitive site-marker displacement results along with computational modeling analysis suggested a preferred location of the GEF binding site as site III, located in subdomain IB of HSA. Some common metal ions have been found to interfere with GEF–HSA interaction.
1. Introduction
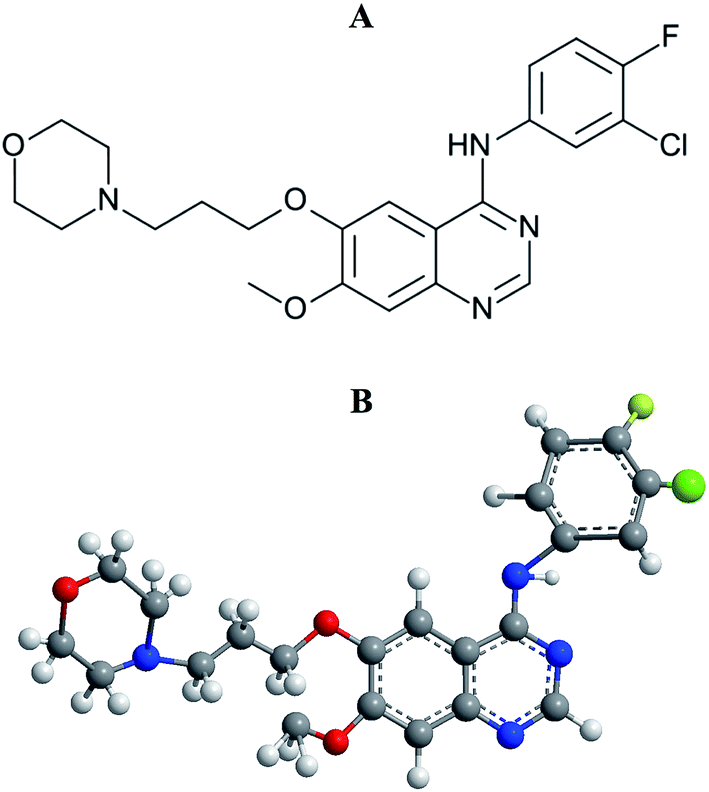
Lung cancer is the most common incidence of cancer worldwide along with leading mortality statistics.1 Due to some undesirable side effects of traditional cancer therapies, a new line of targeted therapeutic agents, i.e., promising anticancer drug molecules being developed for the treatment of various carcinomas.2 Among the wide variety of anticancer drugs used in clinical routine, gefitinib (GEF) is one of the newly Food and Drug Administration (FDA) approved drugs for the treatment of lung cancer.3,4 GEF (Fig. 1) competes with ATP for the ATP-binding site of the epidermal growth factor receptor (EGFR) tyrosine kinase and subsequently freezes the functions of the signaling cascade of the kinase, thus prevents malignancy.3,5 The effective binding of a drug to the protein in blood circulation may influence drug's delivery, distribution, therapeutic efficacy and elimination process.6
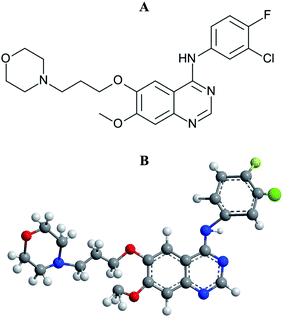 |
| | Fig. 1 Structural representations of gefitinib. (A) 2-D structure and (B) 3-D structure. | |
Human serum albumin (HSA) facilitates the transportation of various drugs through blood circulation and their release at the specific target sites.7,8 Owing to the presence of three well-characterized drug binding sites, viz. sites I, II and III, located in subdomains IIA, IIIA and IB, respectively, of HSA, various drug molecules reversibly bind to the protein, thus leading to an improvement in the drug's pharmacokinetics.7,9,10 Such binding of a drug to the protein also reduces its toxicity and lengthens its in vivo half-life in circulation.11 In view of this, it is important to understand the characteristics of drug–protein interaction.
Various methods are available to investigate the binding of ligands to proteins, which include equilibrium dialysis,12 fluorescence,13 UV-vis absorption and circular dichroism (CD) spectroscopy14 as well as potentiometric titration.15 Among these methods, equilibrium dialysis is widely preferred, which is based on the determination of free and bound ligand concentrations. In many cases, it requires labeled ligands and takes a long time.16 Lack of selectivity of ion selective electrodes for many ligands/drugs limits the use of potentiometric method for ligand binding studies.15 Fluorescence spectroscopic method has been greatly exploited in drug–protein interaction studies in being highly sensitive, less time consuming and easy to perform. Furthermore, information about different binding characteristics such as binding constant, binding stoichiometry, mode of binding and binding mechanism can be obtained from the fluorescence data.17
Although, a recent study has shown the binding characteristics of GEF to bovine serum albumin (BSA),18 no report is available on the binding of GEF to HSA. Due to subtle differences in the amino acid sequence and three-dimensional structure of HSA and BSA,19 drug binding characteristics of these proteins might be different. Since BSA contains two Trp residues compared to one present in HSA, fluorescence quenching data and corresponding results might be different for the two proteins. Furthermore, binding data presented in the previous paper on BSA remains questionable in the absence of inner filter effect correction of the fluorescence data. Therefore, we studied the binding characteristics of GEF to HSA in terms of the binding affinity, interaction forces, location of the binding site and structural changes in HSA upon GEF binding. This paper describes the binding studies of GEF to HSA using fluorescence and circular dichroism spectroscopy along with computational modeling analysis.
2. Materials and methods
2.1. Materials
Human serum albumin (HSA), essentially fatty acid free, indomethacin (IDM), ketoprofen (KTN) and hemin (HMN) were purchased from Sigma-Aldrich Co. (St Louis, MO, USA). Gefitinib (GEF) was a product of Selleckchem (Houston, TX, USA). All other chemicals used were of analytical grade.
2.2. Analytical procedures
The stock protein solution was prepared by dissolving a known amount of HSA in a fixed volume of 60 mM sodium phosphate buffer, pH 7.4 and its concentration was determined spectrophotometrically using a molar extinction coefficient of 36![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 M−1 cm−1 at 280 nm.20
500 M−1 cm−1 at 280 nm.20
The stock solutions (1.0 mg ml−1) of various drugs were prepared by dissolving their crystals in dimethyl sulphoxide (DMSO). These stock solutions were diluted to the desired concentration with the above buffer for experimental use. In all experiments, the final concentration of DMSO in the incubation mixture remained less than 1% (v/v).
All experiments were performed in 60 mM sodium phosphate buffer, pH 7.4 at 25 °C, unless otherwise stated.
2.3. Spectral measurements
2.3.2. CD spectra. Jasco spectropolarimeter (model J-815), equipped with a thermostatically-controlled water-jacketed cell holder was used under constant nitrogen flow to record the CD spectra of protein solutions. The path length of the sample cuvette and the protein concentration used were 1 mm and 3 μM, respectively, for the far-UV CD spectral measurements, while 10 mm path length cuvette and 10 μM protein concentration were employed for CD measurements in the near-UV region. The spectra were recorded using a scan speed of 100 nm min−1 and a response time of 0.5 s. Each spectrum was taken as the average of four successive scans. The measured ellipticity values were expressed in terms of mean residue ellipticity (MRE) in deg. cm2 dmol−1 according to the following equation:| |
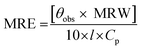 | (1) |
where θobs is the ellipticity in millidegree; MRW is the mean residue weight (molecular weight of the protein, 66500 divided by the total number of amino acids, 585); l is the path length of the cuvette in mm and Cp is the protein concentration in mg ml−1.21
2.3.3. UV-vis absorption spectra. Absorption spectral measurements were made on a UV-vis spectrophotometer (Perkin-Elmer Lambda 25) in the wavelength range, 295–400 nm, using a protein concentration of 3 μM. Absorption spectra of the protein were also obtained in the presence of increasing GEF concentrations (5–50 μM with 5 μM intervals). These data were used for inner filter effect correction.In a separate experiment, absorption spectra of the protein (15 μM) both in the absence and the presence of GEF (5–50 μM with 5 μM intervals) were recorded in the wavelength range, 230–380 nm. Absorption spectra of free GEF solutions of similar concentrations were also obtained in the same wavelength range.
2.4. Ligand binding studies
2.4.1. Fluorescence quenching titration. Interaction of GEF with HSA was studied using fluorescence quenching titration, as described earlier.22 In short, a fixed concentration (3 μM) of HSA was titrated with increasing concentrations of GEF (5–50 μM with 5 μM intervals) in a total volume of 3.0 ml. The fluorescence spectra were recorded in the wavelength range, 310–390 nm upon excitation at 295 nm after 1 h incubation at the desired temperature. The titration was performed at three different temperatures, i.e., 288, 303 and 318 K.The fluorescence data were corrected for the inner filter effect in the same way as described earlier23 using the following equation:
| | |
Fcor = Fobs10(Aex+Aem)/2
| (2) |
where
Fcor and
Fobs refer to the corrected and the observed fluorescence intensity values, while
Aex and
Aem are the differences in the absorbance values of the protein, observed in the presence of ligand at the excitation (295 nm) and the emission (300–400 nm) wavelengths, respectively.
23
2.4.2. Data analysis. To investigate the quenching mechanism involved in the GEF–HSA system, the fluorescence data were treated according to the well-known Stern–Volmer equation:| | |
F0/F = 1 + KSV[Q] = 1 + kqτ0[Q]
| (3) |
where F0 and F refer to the fluorescence intensity values of the protein in the absence and the presence of the quencher (GEF), respectively; [Q] is the concentration of the quencher and KSV is the Stern–Volmer quenching constant.23Values of the bimolecular quenching rate constant (kq) for GEF–HSA system were calculated using the following equation:
where
τ0 is the average lifetime of the fluorophore in the absence of the quencher and it was taken as 6.38 × 10
−9 s for HSA.
24
Values of the binding constant (Ka) for the GEF–HSA system were obtained from the following double logarithmic equation:25
| | |
log(F0 − F)/F = nlogKa − nlog[1/([LT] − (F0 − F)[PT]/F0)]
| (5) |
where
n is the Hill coefficient; [L
T] and [P
T] are the total concentrations of the ligand and the protein, respectively.
Thermodynamic parameters such as the enthalpy change (ΔH), the entropy change (ΔS) and the Gibbs free energy change (ΔG) for the GEF–HSA system were determined to characterize the acting forces involved in the binding process. Values of ΔH and ΔS were obtained from the van't Hoff equation:
| | |
lnKa = −ΔH/RT + ΔS/R
| (6) |
where
R and
T are the gas constant (8.314 J mol
−1 K
−1) and the absolute temperature, respectively. The free energy change, Δ
G of the binding reaction was estimated from the following equation:
2.5. Thermal stability studies
In order to evaluate the effect of GEF binding on the thermal stability of the protein, fluorescence measurements were carried out on HSA (3 μM) in the absence and the presence of GEF (30 μM) in the temperature range, 25–80 °C (with 5 °C intervals). The solution mixture was incubated for 1 h at 25 °C before fluorescence measurements. The fluorescence spectra of HSA and GEF–HSA system were recorded in the wavelength range, 300–400 nm upon excitation at 295 nm. An additional time of 10 min was employed at each temperature for equilibrium establishment.26
2.6. Effect of metal ions on GEF–HSA interaction
To investigate the effect of some common metal ions, i.e., Ba2+, Cu2+, Mn2+, Zn2+, Ca2+, K+ and Mg2+ on the binding of GEF to HSA, titration experiments were performed both in the absence and the presence of metal ions (100 μM) in the same way as described in the Section 2.4.1. An incubation time of 12 h with metal ions was used at 25 °C before fluorescence measurements.
2.7. Competitive site marker displacement experiments
The preferred location of the GEF binding site on HSA was investigated using competitive site marker displacement experiments. Three site markers, used in these experiments were IDM, KTN and HMN for sites I, II and III, respectively.7,10 These experiments were performed by titrating 3 μM HSA and its equimolar complexes with site markers with increasing GEF concentrations (5–50 μM with 5 μM intervals). Site marker–HSA mixtures were allowed to equilibrate for 1 h at room temperature before titration with GEF. Additional 1 h incubation was made after adding GEF and the fluorescence spectra were recorded in the wavelength range, 300–400 nm upon excitation at 295 nm.
2.8. Computational modeling studies
Molecular docking, visualization and rendering simulation were performed using AutoDock 4.2 and AutoDockTools 1.5.6 (ADT).27,28 Structural information of HSA was obtained from the crystal structure (PDB code: 1BM0) with resolution at 2.5 Å. The 3-D coordinates of the protein were retained and all water molecules were removed from the crystal structure of HSA. The atomic coordinates of GEF were extracted from a crystal structure (PDB code: 4I22) and the ligand was set to be flexible during docking analysis with 8 torsional degrees of freedom. Hydrogen atoms were added to GEF and HSA, followed by merging of non-polar hydrogen atoms for efficient computation. Furthermore, gasteiger partial charges were computed and assigned to all atoms. For each binding site (sites I, II and III), an independent docking analysis of 100 runs was performed within a grid box with 70 × 70 × 70 grid points and a grid space of 0.375 Å. The coordinates of center of grid box were at x = 41.61, y = 33.78 and z = 30.49 for binding site I; x = 11.61, y = 29.78 and z = 18.49 for binding site II and x = 46.61, y = 22.78 and z = 14.49 for binding site III. The search method used was Lamarckian genetic algorithms with 250000 energy evaluations. Cluster analysis (RMSD tolerance at 2.0 Å) and docking result were examined using AutoDockTools 4.2. Chimera 1.10.2 software was employed to visualize the GEF–HSA complex.29
2.9. Statistical analysis
All data were expressed as the average ± standard deviation (SD) from a minimum of three experiments. The curves plotting and statistical data processing were made using the OriginPro 8.5 software (OriginLab Corp., Northampton, MA).
3. Results and discussion
3.1. GEF–HSA interaction
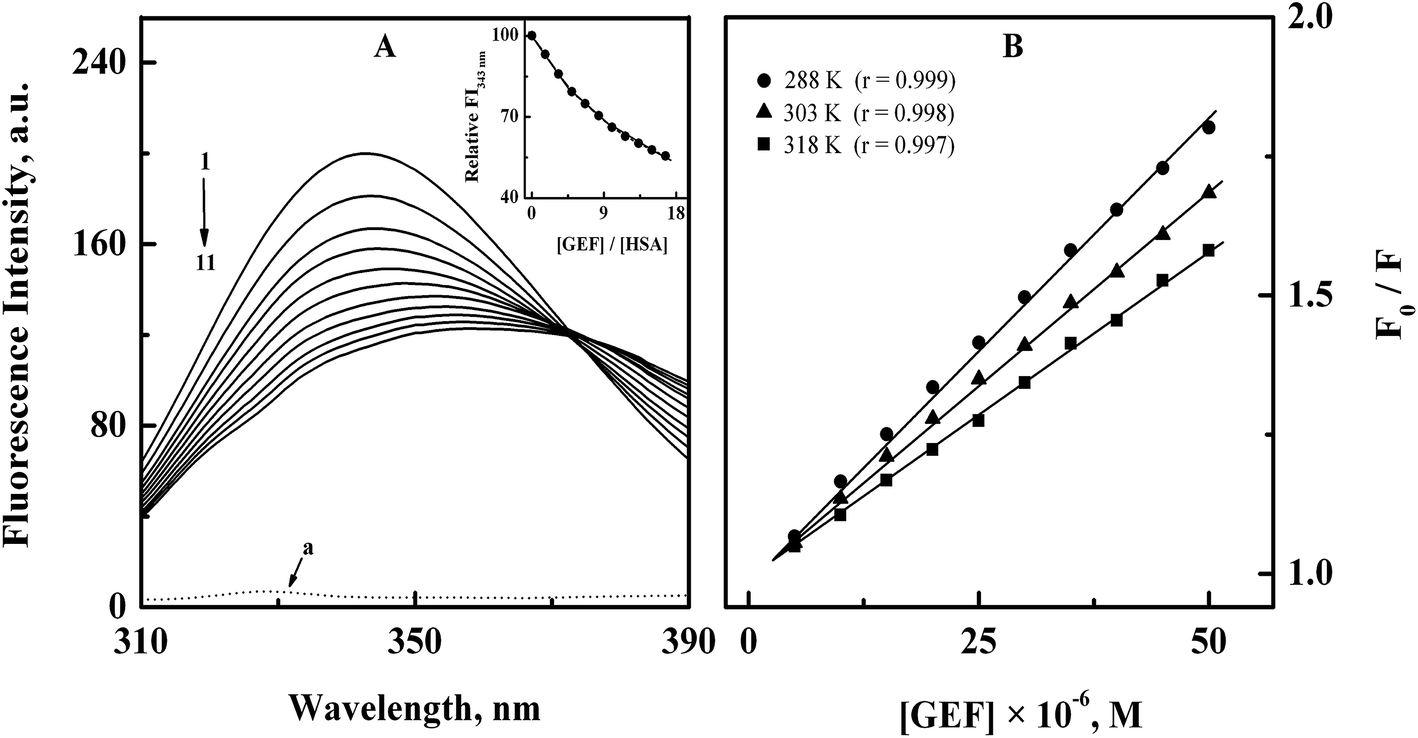
3.1.1. Fluorescence spectra and quenching mechanism. The intrinsic fluorescence of the protein is mostly contributed by its aromatic fluorophores, tryptophan (Trp) and tyrosine (Tyr) residues,30 which may be affected by the binding of a ligand to the protein.31–33 The fluorescence spectrum of HSA showed an emission maximum at 343 nm (Fig. 2A) due to the presence of lone tryptophan (Trp-214) residue at subdomain IIA of HSA.34 Addition of increasing GEF concentrations led to significant quenching in the protein's fluorescence along with red shift in the emission maximum in a concentration dependent manner (Fig. 2A). It is important to note that free GEF did not produce any fluorescence in this range (spectrum ‘a’). About 45% quenching in the fluorescence intensity (inset of Fig. 2A) and 15 nm red shift in the emission maximum, observed at the highest GEF concentration (50 μM) clearly suggested the binding of GEF to HSA. Occurrence of the red shift in the emission maximum can be ascribed to the change in the microenvironment around lone Trp residue from nonpolar to polar.35 On the other hand, a variety of molecular processes such as excited-state reactions, molecular rearrangements, energy transfer, ground-state complex formation and collisional quenching may be responsible for the observed quenching of the protein fluorescence upon ligand binding.23 Several earlier reports have shown quenching of HSA fluorescence upon drug binding with red shift in the emission maximum.36–38
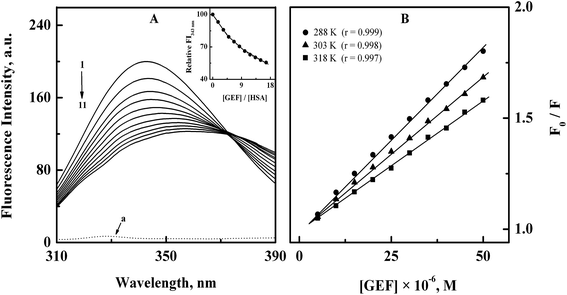 |
| | Fig. 2 (A) Fluorescence quenching spectra of 3 μM HSA (spectrum 1) with increasing concentrations (5–50 μM with 5 μM intervals) of GEF (spectra 2–11), obtained in 60 mM sodium phosphate buffer, pH 7.4 at 15 °C upon excitation at 295 nm. The dotted line at the bottom (a) shows the fluorescence spectrum of GEF alone. Inset displays the decrease in the relative fluorescence intensity of HSA at 343 nm (FI343 nm) with increasing GEF/HSA molar ratios. (B) Stern–Volmer plots for the binding of GEF to HSA, obtained at three different temperatures, i.e., 288, 303 and 318 K. | |
Analysis of the fluorescence quenching data obtained at three different temperatures (288, 303 and 318 K) according to the eqn (3) yielded the Stern–Volmer plots, as shown in Fig. 2B. These plots exhibited good linearity with a correlation coefficient (r) ≥ 0.997 throughout the GEF concentrations used. Values of KSV were obtained from the linear regression analysis of the above plots and are listed in Table 1. Two types of the fluorescence quenching phenomena, namely, static and dynamic quenching can be differentiated by KSV dependence on temperature. The quenching constant is expected to decrease with increasing temperature for static quenching, while the reverse trend characterizes dynamic quenching.23,39 As shown in Table 1, value of KSV significantly decreased with increasing temperature, thus indicating the characteristic of static quenching. Similar trend of KSV with increasing temperature was observed in a previous study on GEF–BSA interaction.18 Therefore, the observed quenching in the protein's fluorescence upon GEF addition seems to be the result of GEF–HSA complex formation.
Table 1 Binding constants and thermodynamic parameters for GEF–HSA interaction, studied at three different temperatures, pH 7.4
| T (K) |
KSV (M−1) |
Ka (M−1) |
ΔS (J mol−1 K−1) |
ΔH (kJ mol−1) |
ΔG (kJ mol−1) |
| 288 |
(1.60 ± 0.03) × 104 |
(1.70 ± 0.07) × 104 |
|
|
−23.32 |
| 303 |
(1.36 ± 0.07) × 104 |
(1.43 ± 0.04) × 104 |
+54.06 |
–7.74 |
−24.13 |
| 318 |
(1.19 ± 0.06) × 104 |
(1.25 ± 0.08) × 104 |
|
|
−24.94 |
Furthermore, kq values, viz., 2.51 × 1012, 2.13 × 1012 and 1.87 × 1012 M−1 s−1 obtained at 288, 303 and 318 K, respectively, for the GEF–HSA system were significantly higher than the value of 2 × 1010 M−1 s−1, reported for the maximum dynamic quenching constant for the association of various quenchers and the fluorophore in a bimolecular complex.40 Shen and coworkers18 have also reported similar order of magnitude (1012) for kq values in GEF–BSA system. Therefore, it can be concluded that the quenching mechanism involved in the GEF–HSA system was initiated by static rather than dynamic quenching process.
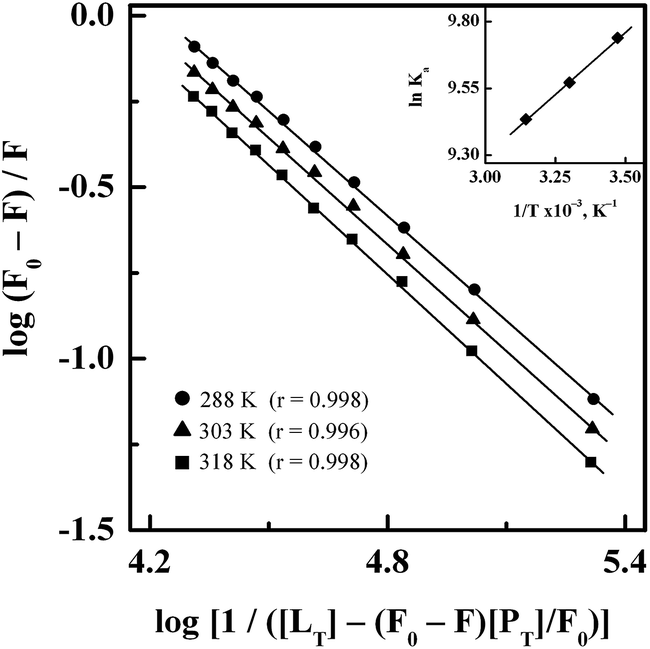
3.1.2. Binding affinity. Fig. 3 shows double logarithmic plots for the binding of GEF to HSA at different temperatures, as obtained after treatment of the fluorescence quenching data according to eqn (5). Values of the binding constant (Ka) for the GEF–HSA system at three different temperatures were retrieved from these plots by dividing the Y-axis intercept with the slope value and are included in Table 1. Since the Ka value for the GEF–HSA system was found to remain in the range of 1.70–1.25 × 104 M−1, it indicated a moderate binding affinity between GEF and HSA. BSA has been found to bind GEF with slightly higher affinity (Ka = 6.61 × 104 M−1 at 25 °C).18 Such moderate binding affinity is beneficial for the efficient transport of the drug and its subsequent release at its target site. As can be seen from Table 1, the Ka value of the GEF–HSA system showed a decreasing trend with increasing temperature due to decomposition of the GEF–HSA complex at higher temperature. Several published reports on the binding of various drugs to HSA have shown moderate affinity.33,41,42
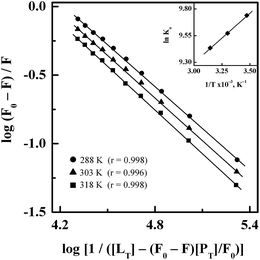 |
| | Fig. 3 Double logarithmic plots of log(F0 − F)/F versus log[1/([LT] − [(F0 − F)[PT]/F0)] for the binding of GEF to HSA, obtained at three different temperatures, i.e., 288, 303 and 318 K. Inset shows the van't Hoff plot for the GEF–HSA system. | |
3.1.3. Interaction forces. In view of the temperature dependence of the binding constant, ligand–protein interaction seems to be a thermodynamic process.32 Therefore, determination of thermodynamic parameters, i.e., ΔS, ΔH and ΔG for GEF–HSA interaction is important to predict the acting forces involved in the binding reaction. The inset of Fig. 3 shows linear van't Hoff plot for GEF–HSA interaction. Values of ΔH and ΔS, as obtained from the slope and the intercept, respectively, of the van't Hoff plot along with the ΔG values at three different temperatures are listed in Table 1. The negative sign of ΔG value showed spontaneous nature of the binding reaction at all temperatures. In addition, the negative value of ΔH revealed that the formation of GEF–HSA complex was an exothermic process.Various noncovalent forces, such as hydrophobic interactions, hydrogen bonds, van der Waals interactions and electrostatic interactions are known to stabilize ligand–protein complexes.43,44 The sign and magnitude of ΔS and ΔH are useful in predicting the nature of the forces involved in various ligand–protein binding processes.45 For example, a positive vale of ΔS is regarded as an evidence for hydrophobic interactions, while the hydrogen bonding as well as van der Waals interactions are accompanied by a negative ΔH value.45 In view of the positive ΔS value and negative ΔH value, obtained for GEF–HSA system, hydrophobic interactions along with hydrogen bonds and van der Waals forces seem to favor the stabilization of GEF–HSA complex. A previous study has reported the involvement of van der Waals interactions and hydrogen bonds in the binding of GEF to BSA.18 Participation of similar forces in the binding of GEF to HSA and BSA is not surprising as both BSA and HSA show 76% structural similarity.46 Several published reports have shown the involvement of hydrophobic interactions and hydrogen bonds in ligand–protein association process, based on the positive ΔS and negative ΔH values.22,47,48 Although, electrostatic interactions are also accompanied by a positive ΔS value, but the value of ΔH has been found to be either small or close to zero.45 Accordingly, the large negative value of ΔH (−7.74 kJ mol−1) for GEF–HSA system cannot be taken as an evidence for the electrostatic interactions.45,47 Absence of charged group in the GEF molecule and negative ΔH value, obtained in this study excluded the participation of electrostatic interactions in the stabilization GEF–HSA complex.45 On the other hand, it is feasible to account more than one intermolecular binding forces for ligand–protein interactions on the basis of thermodynamic parameters.47 Thus, thermodynamic data clearly revealed both hydrophobic interactions and hydrogen bonds as the major stabilizing forces in the GEF–HSA complex formation. This was further supported by our molecular docking results, as described in the Section 3.7.
3.2. UV-vis absorption results
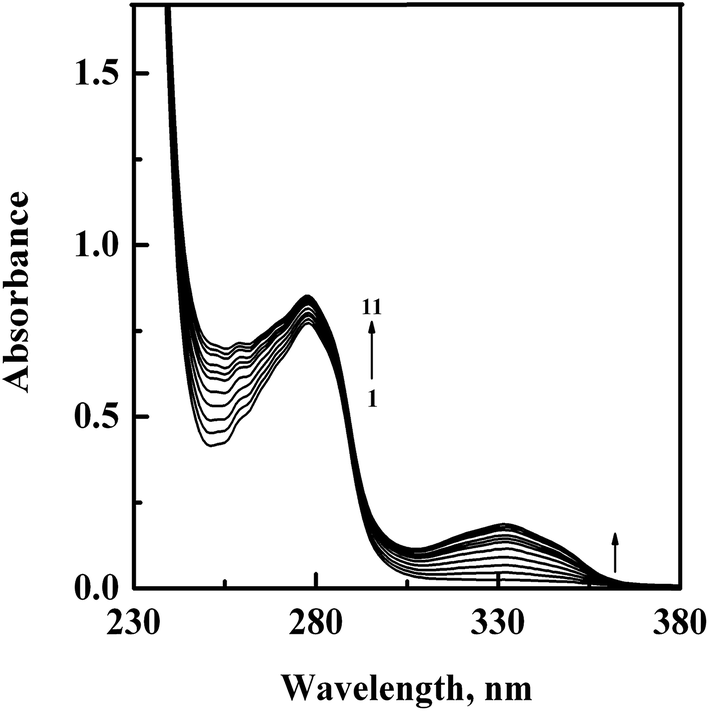
In order to confirm the complex formation between GEF and HSA, UV-vis absorption spectra of HSA were studied in the absence (spectrum 1) and presence (spectra 2–11) of increasing GEF concentrations (Fig. 4). These changes in the UV absorption spectra of the protein at respective GEF concentrations were obtained by subtracting the spectra of the pure GEF solutions from the spectra of GEF–HSA mixtures (ESI Fig. 1A and B†). Significant change in the absorbance value of HSA at 280 nm upon GEF addition suggested microenvironmental perturbations around the protein chromophores due to the complex formation between GEF and HSA. Absorption spectrum of free GEF was characterized by the presence of a peak at 332 nm (ESI Fig. 1B†). Increase in the absorbance value at 332 nm with increasing GEF concentration was also noticed, which was suggestive of the complex formation between GEF and HSA. Such changes in the absorption spectrum of HSA in the presence of GEF supported the involvement of static quenching mechanism in GEF–HSA system.
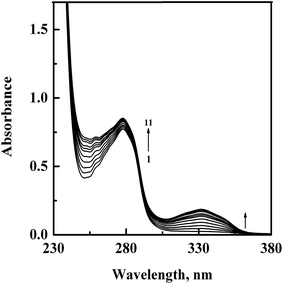 |
| | Fig. 4 UV-vis absorption spectra of HSA (15 μM), obtained in the absence (spectrum 1) and presence (spectra 2–11) of increasing GEF concentrations (5–50 μM with 5 μM intervals) in 60 mM sodium phosphate buffer, pH 7.4 at 25 °C. | |
3.3. Ligand-induced microenvironmental perturbations around protein fluorophores
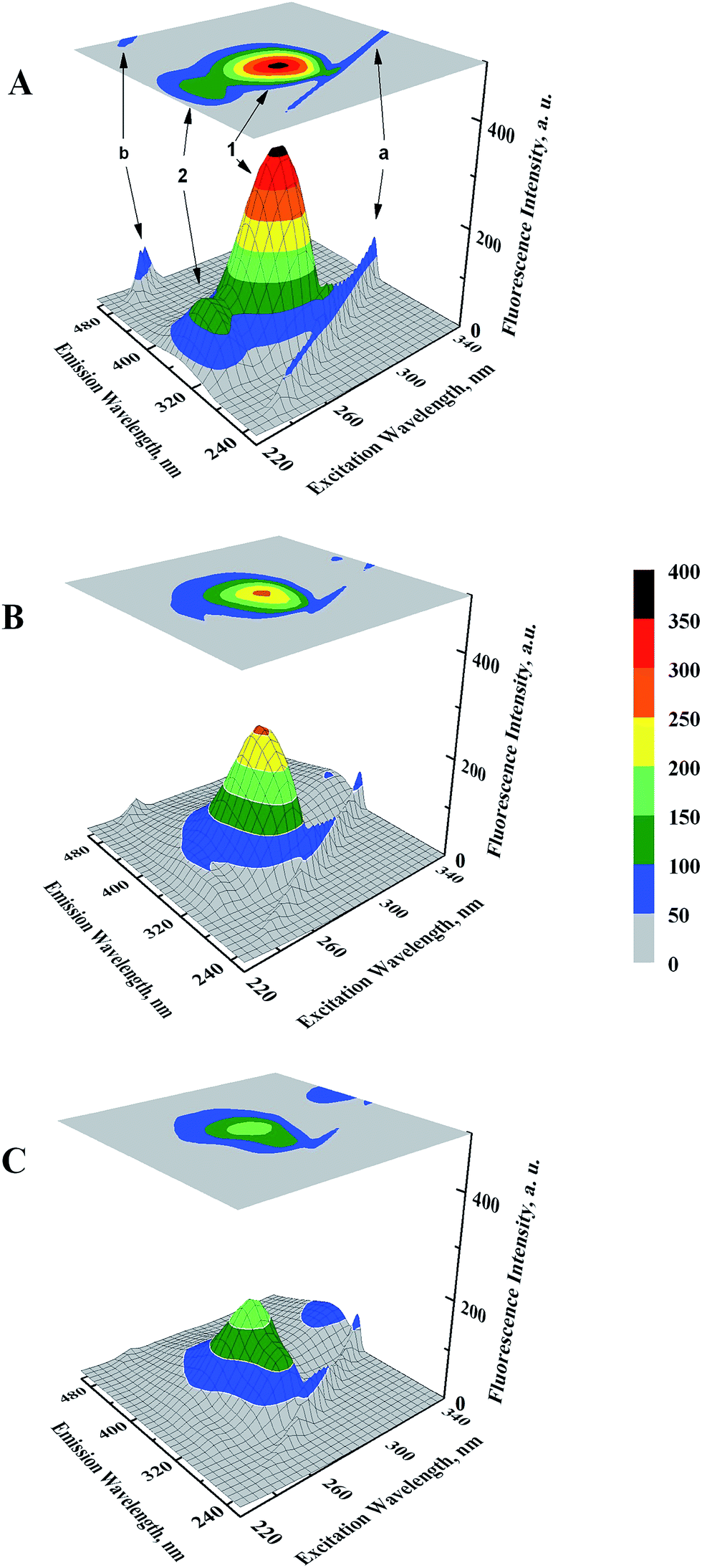
Microenvironmental perturbations around Tyr and Trp residues of the protein induced by ligand binding can be observed by studying the three-dimensional fluorescence spectral changes in HSA in the presence of GEF. The 3-D fluorescence spectra and corresponding contour maps of HSA (A) and GEF–HSA systems (B and C) are shown in Fig. 5, while spectral characteristics are listed in Table 2. Four peaks are commonly observed in the 3-D fluorescence spectrum of HSA. Peaks ‘a’ and ‘b’ are known as the first-order Rayleigh scattering peak (λex = λem) and the second-order Rayleigh scattering peak (2λex = λem), respectively.23,30 In addition, two strong fluorescence peaks, viz. peaks 1 and 2 were due to spectral characteristics of Tyr and Trp residues of HSA. A comparison of the 3-D fluorescence spectral characteristics of HSA in the absence and the presence of GEF (5 molar excess) showed ∼28% reduction in the intensity along with 16 nm red shift in the emission maximum of peak 1 and ∼76% reduction in the intensity along with 12 nm red shift in peak 2 (Table 2). These changes in the fluorescence characteristics of the peaks became more pronounced in the presence of 10 molar excess of GEF (Table 2). Such changes in the fluorescence spectral characteristics clearly indicated microenvironmental perturbation (from nonpolar to polar) around Trp and Tyr residues in subdomain IIA of HSA, which housed the lone Trp-214 of HSA.
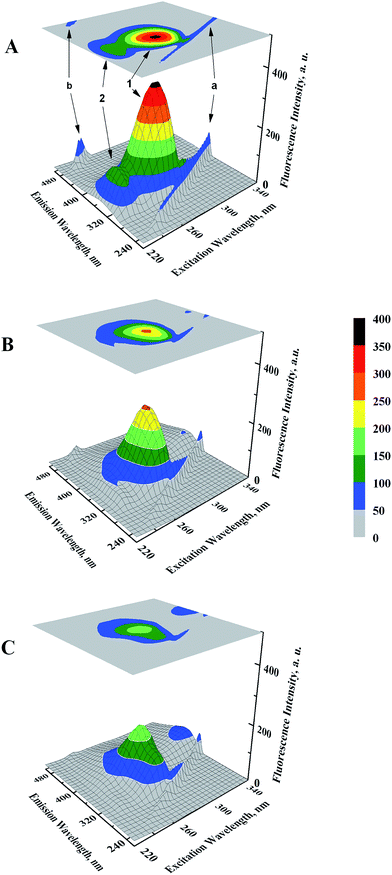 |
| | Fig. 5 Three-dimensional fluorescence spectral projections and corresponding contour maps of (A) 3 μM HSA, (B) GEF–HSA (5:1) and (C) GEF–HSA (10:1) systems, obtained in 60 mM sodium phosphate buffer, pH 7.4 at 25 °C. | |
Table 2 Three-dimensional fluorescence spectral characteristics of HSA (3 μM) and GEF–HSA complexes, obtained at 25 °C, pH 7.4
| System |
Peak |
Peak position [λex/λem (nm/nm)] |
Intensity |
| HSA |
 |
230/230 → 350/350 |
16.44 → 88.91 |
| 250/500 |
89.28 |
| 280/337 |
361.02 |
| 230/335 |
114.41 |
| [GEF]:[HSA] = 5:1 |
 |
230/230 → 350/350 |
14.70 → 75.92 |
| 250/500 |
40.55 |
| 280/353 |
261.10 |
| 230/347 |
27.74 |
| [GEF]:[HSA] = 10:1 |
 |
230/230 → 350/350 |
14.81 → 71.06 |
| 250/500 |
22.95 |
| 280/361 |
190.73 |
| 230/361 |
19.04 |
3.4. Ligand-induced structural changes
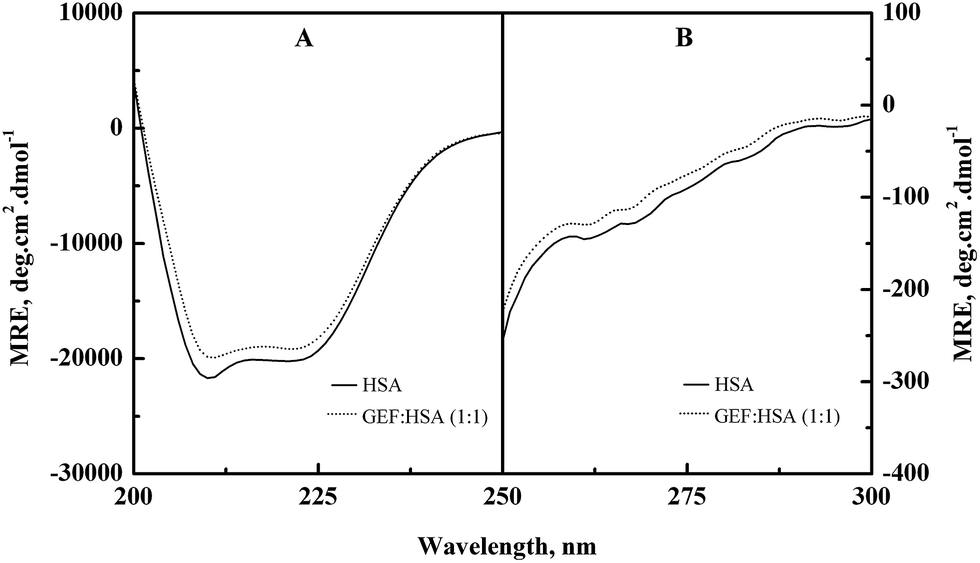
Alterations in the secondary and the tertiary structures of HSA in the presence of GEF were evident from the far-UV (Fig. 6A) and the near-UV (Fig. 6B) CD spectra of the protein, respectively. Presence of the α-helical structure in HSA was reflected from the appearance of two minima at 208 and 222 nm in the far-UV CD spectra (Fig. 6A). Binding of GEF to HSA in 1:1 molar ratio induced significant change in the far-UV CD spectrum of HSA (Fig. 6A), thus suggesting secondary structural changes in the protein.
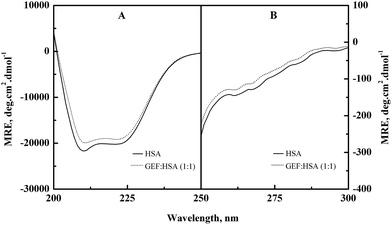 |
| | Fig. 6 (A) Far-UV and (B) near-UV CD spectra of HSA in the absence and the presence of equimolar concentration of GEF, obtained in 60 mM sodium phosphate buffer, pH 7.4 at 25 °C. The CD spectra were recorded using a protein concentration of 3 μM and 10 μM in the far-UV and the near-UV regions, respectively. | |
The near-UV CD spectrum of HSA was characterized by the spectral features due to aromatic chromophores and disulfide bonds present in the protein. Appearance of two minima around 263 and 269 nm and shoulders around 282 and 291 nm characterized the near-UV CD spectrum of HSA. Significant alteration in the near-UV CD spectra of HSA was observed in the presence of GEF, suggesting tertiary structural changes in HSA due to GEF binding.
Similar changes in the far- and near-UV CD spectra were also noticed in BSA in the presence of GEF.18
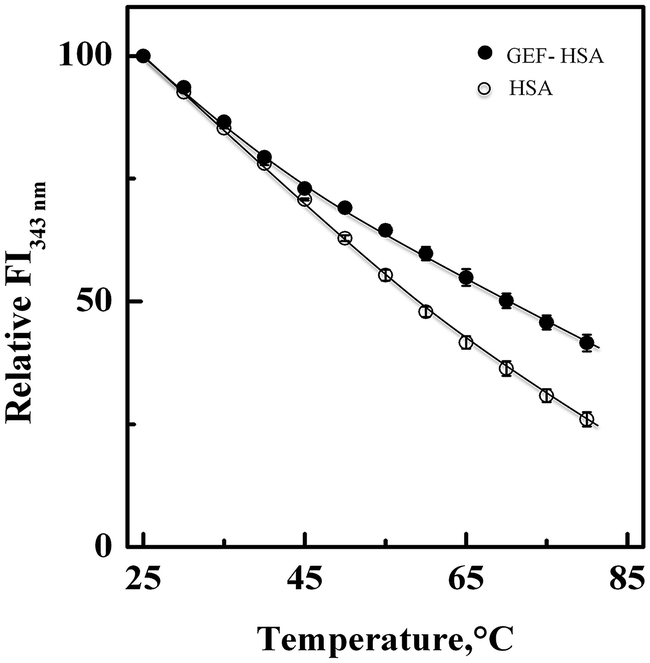
3.5. Thermal stabilization of HSA upon GEF binding
Fig. 7 shows the influence of temperature on the fluorescence intensity at 343 nm (FI343 nm) of HSA as well as GEF–HSA system in the temperature range, 25–80 °C. HSA showed a gradual decrease in FI343 nm with the increase in temperature. Interestingly, lesser decrease in the FI343 nm was noticed in the presence of GEF at higher temperatures (>45 °C) compared to that observed in its absence. These results clearly suggested significant protection of HSA against temperature-induced structural changes at higher temperatures. Such changes in the fluorescence intensity can be explained by coupling of binding and unfolding equilibria.49,50 Several earlier reports have shown ligand-induced thermal stabilization of HSA.51,52
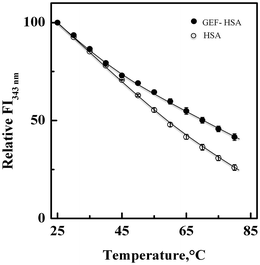 |
| | Fig. 7 Effect of temperature on the fluorescence intensity of HSA at 343 nm (FI343 nm) in the absence and the presence of GEF, studied in the temperature range, 25–80 °C. The spectra were obtained with 3 μM HSA and GEF–HSA (10:1) system in 60 mM sodium phosphate buffer, pH 7.4. | |
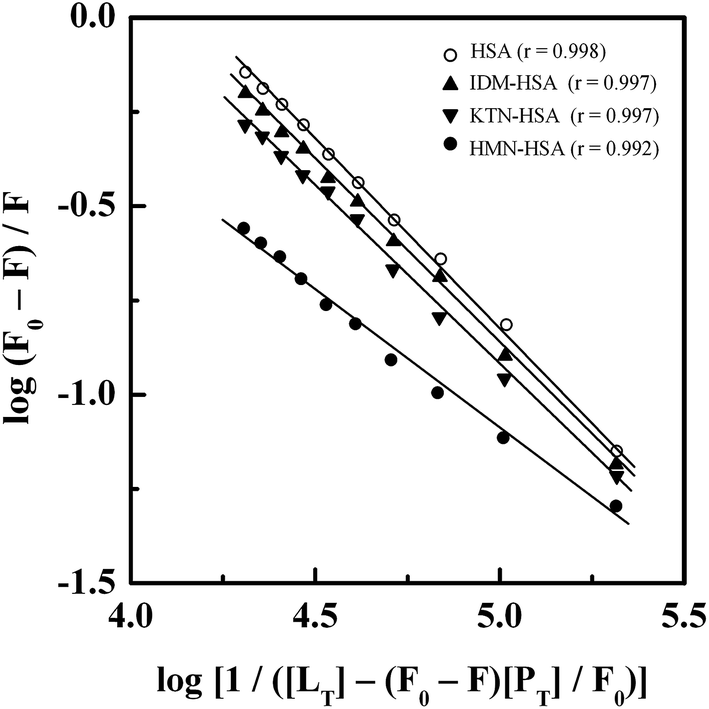
3.6. Location of the GEF binding site
Treatment of the fluorescence quenching titration results of HSA and its complexes with different site markers with increasing GEF concentrations according to eqn (5) yielded the double logarithmic plots, as shown in Fig. 8. In order to compare the influence of site markers on the binding of GEF to HSA, values of the binding constant (Ka), obtained in the absence and the presence of site markers were determined (Table 3). As can be seen from the Table 3, decrease in the Ka value for GEF–HSA interaction was significantly higher in the presence of HMN, compared to those observed with KTN/IDM-bound HSA. In other words, presence of HMN, which is known to bind to site III, located in subdomain IB, significantly affected the binding of GEF to HSA. These results clearly suggested site III as the preferred GEF binding site on HSA. Furthermore, these results were in line with our molecular docking analysis, as discussed in the Section 3.7. Site I (subdomain IIA) has been proposed as the GEF binding site in BSA based on competitive ligand displacement experiments.18 However, binding site III was not explored in the previous study.18 Furthermore, a decrease in the logKb value from 4.92 to 4.58 has been taken as the evidence for site I selection.18
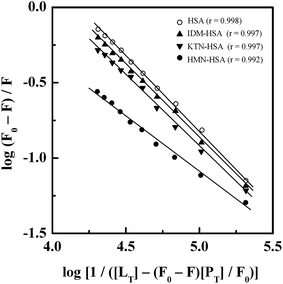 |
| | Fig. 8 Double logarithmic plots of log(F0 − F)/F versus log[1/([LT] − [(F0 − F)[PT]/F0)] for the GEF–HSA system in the absence and the presence of different site markers, i.e., IDM, KTN and HMN, obtained at 25 °C, pH 7.4. | |
Table 3 Binding constant for GEF–HSA interaction in the absence and the presence of site markers, obtained at 25 °C, pH 7.4
| Site markers |
Ka (× 104 M−1) |
| — |
1.53 ± 0.04 |
| IDM |
1.35 ± 0.08 |
| KTN |
1.10 ± 0.07 |
| HMN |
0.29 ± 0.03 |
3.7. Computational modeling analysis
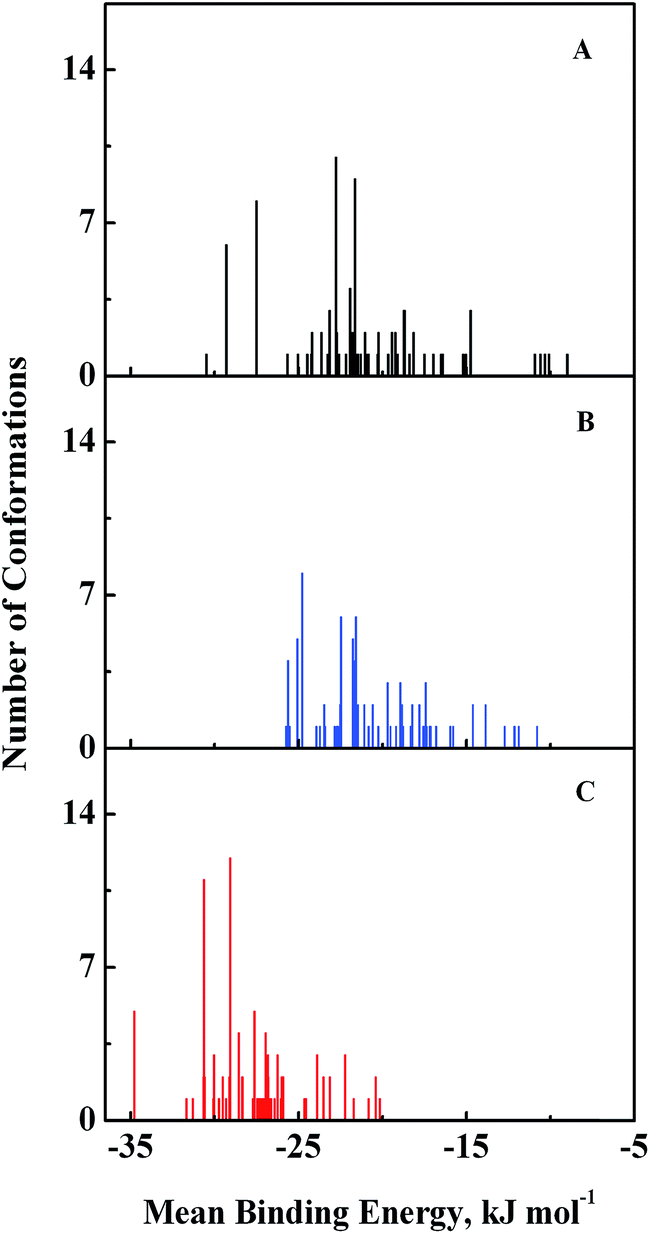
Computational modeling analysis allows elucidation of the most favored binding mode of ligand to binding site at atomic resolution. In docking analysis, GEF was set to be flexible with 8 torsional degrees of freedom due to rotatable bonds. Estimated free energy of binding was computed for each binding mode based on a semi-empirical force field with evaluated energy terms such as electrostatic interaction, torsional entropy, hydrophobic interaction and others. The energy minimized and geometrically optimized ligand was allowed to explore the configuration space within grid boxes centered at drug binding sites I, II and III of HSA. For each binding site, we performed 100 rounds of docking followed by ranking of binding modes based on their estimated free energy of binding. Binding modes of GEF were clustered according to conformational similarity using root mean square deviation (RMSD) values with cutoff RMSD at 2.0 Å. Comparison of binding modes in three binding sites showed gefitinib binds more favorably to binding site III (Fig. 9). As shown in Fig. 9, 10 clusters constituted by 40 out of 100 binding modes exhibit mean binding energy lower than −29.3 kJ mol−1 in binding site III. However, most of the binding mode clusters in binding sites I and II possesses mean binding energy > −29.3 kJ mol−1. Therefore, the binding site III of HSA was the preferred binding site of GEF, as suggested by cluster analysis.
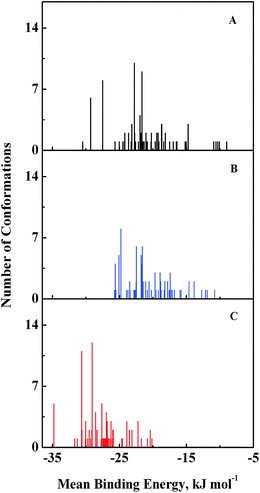 |
| | Fig. 9 Cluster analysis of the docking of GEF to three ligand binding sites, viz., I (A), II (B) and III (C) of HSA crystal structure, 1BM0. A total of 100 runs were performed for each binding site. | |
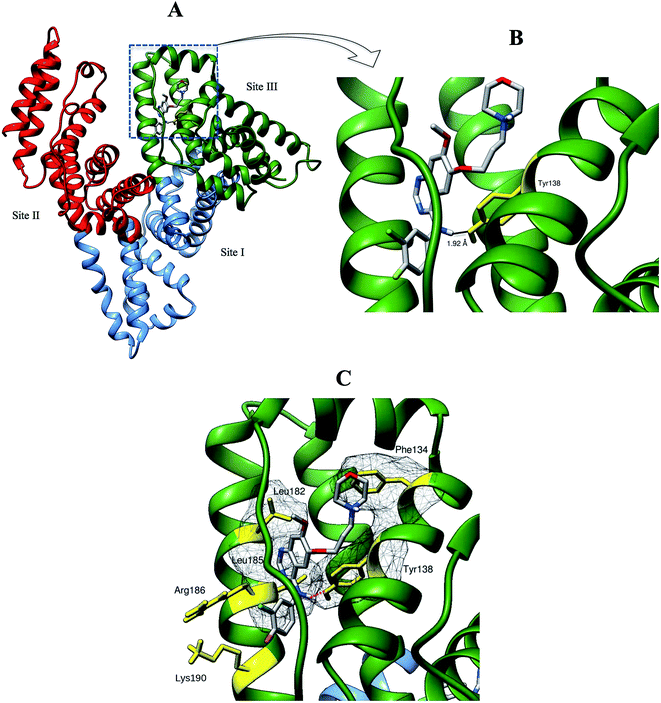 |
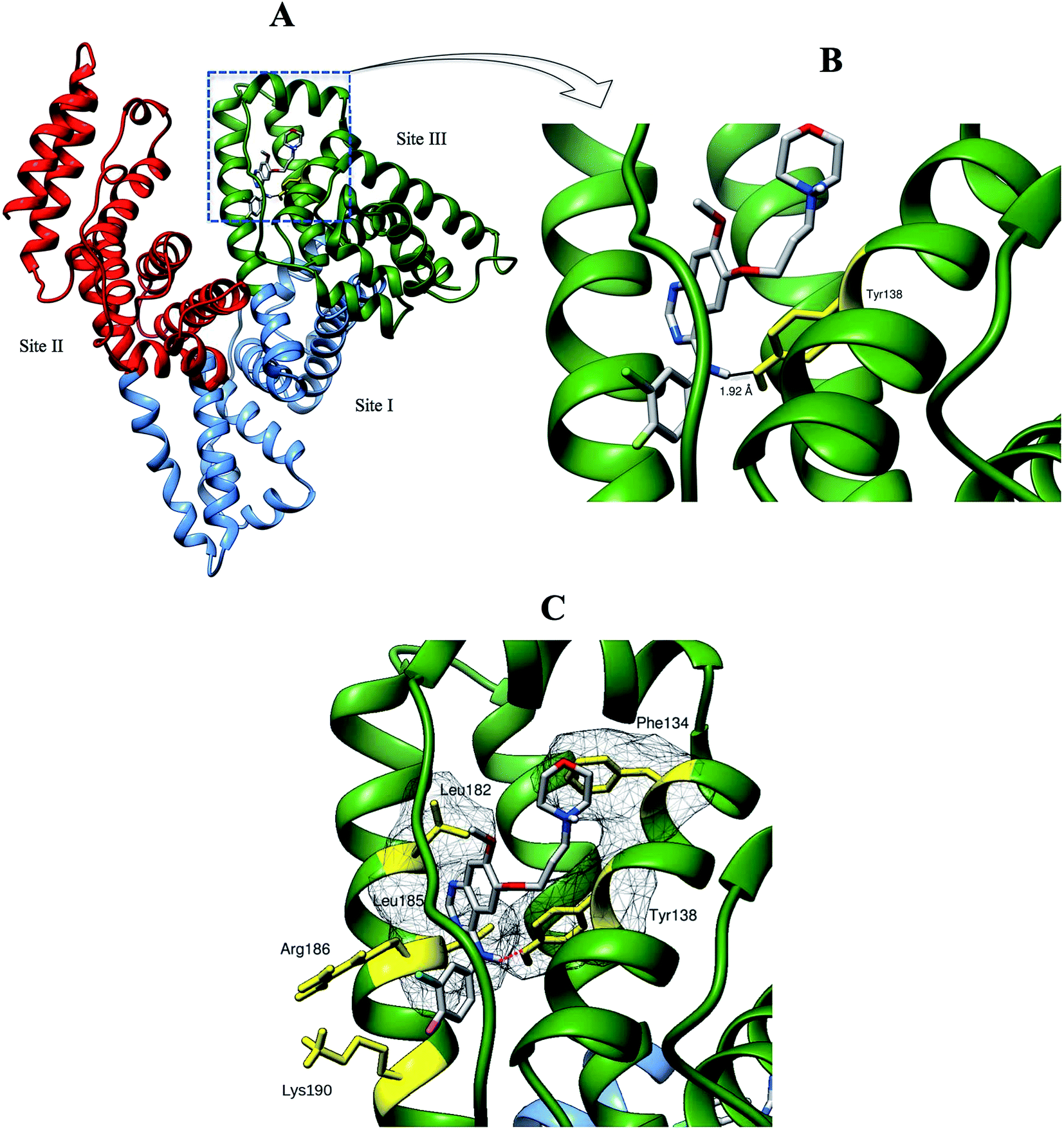
| | Fig. 10 (A) Binding orientations of the lowest docking energy conformation of GEF (rendered in sticks) in the subdomain IB (binding site III) of HSA. Different domains of HSA are shown in green (domain I), sky blue (domain II) and red (domain III) colors. (B) The enlarged view of the binding locus showing a hydrogen bond (black line) between Tyr-138 (rendered in yellow stick) and GEF. (C) Residues involved in hydrophobic interactions are shown with their molecular surface in mesh surface representation. Interacting residues with their side chains are colored in yellow while red dashed line indicates hydrogen bond. | |
We selected the best-scored binding mode from the cluster with the lowest binding energy in binding site III for subsequent analyses (Fig. 10). The binding energy of GEF docked to binding site III is computed to be at −36.4 kJ mol−1. At the binding site III, GEF docked to a hydrophobic pocket walled by 21 amino acids within 5 Å: Leu-115, Val-116, Arg-117, Pro-118, Met-123, Phe-134, Lys-137, Tyr-138, Glu-141, Ile-142, His-146, Phe-149, Phe-157, Tyr-161, Lys-181, Leu-182, Asp-183, Leu-185, Arg-186, Gly-189 and Lys-190. Hydrophobic interaction would be a major factor that stabilizes the complex of GEF–HSA through Phe-134, Tyr-138, Leu-182 and Leu-185 in proximity (Fig. 10C). However, the interaction between GEF and HSA cannot be presumed to be exclusively hydrophobic in nature; as there were several polar residues in the proximity of the bound ligand that may participate in polar interactions with the hydrophilic groups of GEF. One hydrogen bond was predicted between hydroxyl group of Tyr-138 and amine group of GEF with bond distance of 1.92 Å (Fig. 10B). Therefore, our docking simulation predicted that GEF has a binding preference for binding site III, located in subdomain IB of HSA and involves both hydrophobic interactions and hydrogen bonds in the GEF–HSA complex formation. These docking results were consistent with our competitive site marker displacement results as shown in the Section 3.6. In a previous study, docking analysis with BSA revealed the preferred binding location of GEF in subdomain IIA (site I) of BSA.18 In the absence of the docking results for site III, conclusions about site I as the preferred GEF binding site remains questionable.
3.8. Metal ions interference with GEF–HSA interaction
Presence of some common metal ions, viz., Ba2+, Cu2+, Mn2+, Zn2+, Ca2+, K+ and Mg2+ in the blood plasma might affect the binding of a drug to the protein.26 The interference of these common ions with GEF–HSA interaction was investigated by determining the Ka values of GEF–HSA binding reaction in the absence and the presence of these metal ions. As shown in Table 4, both increase and decrease in the Ka value was noticed in the presence of these metal ions. Whereas, K+ and Mg2+ produced slight increase in the Ka value, decrease in the Ka was observed in the presence of Ba2+, Cu2+, Mn2+ and Zn2+ ions. These results clearly suggested some influence of metal ions on the binding of GEF to HSA, which may prolong and/or weaken the storage time of the drug in plasma. Therefore, it is important to adapt the dose of the drug in the presence of these ions to achieve the desired therapeutic effect.32,53
Table 4 Binding constant for GEF–HSA interaction at 25 °C, pH 7.4 in the absence and the presence of some common ions
| Metal ions |
Ka (× 104 M−1) |
| — |
1.53 ± 0.04 |
| Ba2+ |
0.79 ± 0.08 |
| Cu2+ |
1.02 ± 0.07 |
| Mn2+ |
1.08 ± 0.09 |
| Zn2+ |
1.16 ± 0.10 |
| Ca2+ |
1.69 ± 0.08 |
| K+ |
1.91 ± 0.13 |
| Mg2+ |
2.10 ± 0.17 |
4. Conclusions
Molecular characterization of the binding of GEF to HSA was made in terms of the binding affinity (Ka = 1.53 × 104 M−1 at 25 °C), thermodynamic data (ΔH = −7.74 kJ mol−1 and ΔS = +54.06 J mol−1 K−1), interaction forces involved (hydrophobic interactions and hydrogen bonds), change in the protein's secondary and tertiary structures as well as microenvironmental perturbation around protein fluorophores upon drug binding. GEF binding to HSA improved thermal stability of the protein and site III (subdomain IB) was identified as the GEF binding site on HSA. These findings provided detailed insight to understand the binding properties of GEF to HSA, which may be beneficial for medical and pharmaceutical applications in future.
Acknowledgements
This work was financially supported by the High Impact Research MoE Grant UM.C/625/1/HIR/MoE/SC/02, sanctioned by the Ministry of Education, Government of Malaysia and the University of Malaya. Md. Zahirul Kabir gratefully acknowledges the financial assistance from the University of Malaya in the form of doctoral fellowship under the Bright Sparks Program (BSP/APP/1892/2013). The authors thank the Dean, Faculty of Science and the Head, Institute of Biological Sciences, University of Malaya for providing necessary facilities.
References
- B. W. Stewart and C. P. Wild, World Cancer Report 2014, World Health Organization, 2014 Search PubMed.
- I. M. Shih and T. L. Wang, Notch signaling, γ-secretase inhibitors, and cancer therapy, Cancer Res., 2007, 67, 1879–1882 CAS.
- E. Raymond, S. Faivre and J. P. Armand, Epidermal growth factor receptor tyrosine kinase as a target for anticancer therapy, Drugs, 2000, 60, 15–23 CAS.
- K. Yanase, S. Tsukahara, S. Asada, E. Ishikawa, Y. Imai and Y. Sugimoto, Gefitinib reverses breast cancer resistance protein-mediated drug resistance, Mol. Cancer Ther., 2004, 3, 1119–1125 CAS.
- M. Ono and M. Kuwano, Molecular mechanisms of epidermal growth factor receptor (EGFR) activation and response to gefitinib and other EGFR-targeting drugs, Clin. Cancer Res., 2006, 12, 7242–7251 CrossRef CAS PubMed.
- R. E. Olson and D. D. Christ, Plasma protein binding to drugs, Annu. Rep. Med. Chem., 1996, 31, 327–336 CAS.
- U. Kragh-Hansen, V. T. G. Chuang and M. Otagiri, Practical aspects of the ligand-binding and enzymatic properties of human serum albumin, Biol. Pharm. Bull., 2002, 25, 695–704 CAS.
- E. Lazaro, P. J. Lowe, X. Briand and B. Faller, New approach to measure protein binding based on a parallel artificial membrane assay and human serum albumin, J. Med. Chem., 2008, 51, 2009–2017 CAS.
- G. Sudlow, D. J. Birkett and D. N. Wade, The characterization of two specific drug binding sites on human serum albumin, Mol. Pharmacol., 1975, 11, 824–832 CAS.
- U. Kragh-Hansen, Relations between high-affinity binding sites of markers for binding regions on human serum albumin, Biochem. J., 1985, 225, 629–638 CAS.
- U. Kragh-Hansen, Molecular and practical aspects of the enzymatic properties of human serum albumin and of albumin–ligand complexes, Biochim. Biophys. Acta, 2013, 1830, 5535–5544 CrossRef CAS PubMed.
- M. Tanaka, Y. Asahi and S. Masuda, Interaction between drugs and water-soluble polymers VII. Binding of berberine with bovine serum albumin, J. Macromol. Sci., Part A: Pure Appl. Chem., 1995, 32, 339–347 Search PubMed.
- S. Tunc, O. Duman and B. K. Bozoglan, Studies on the interactions of chloroquine diphosphate and phenelzine sulfate drugs with human serum albumin and human hemoglobin proteins by spectroscopic techniques, J. Lumin., 2013, 140, 87–94 CAS.
- O. Duman, S. Tunc and B. K. Bozoglan, Characterization of the binding of metoprolol tartrate and guaifenesin drugs to human serum albumin and human hemoglobin proteins by fluorescence and circular dichroism spectroscopy, J. Fluoresc., 2013, 23, 659–669 CrossRef CAS PubMed.
- E. Ayranci and O. Duman, Binding of lead ion to bovine serum albumin studied by ion selective electrode, Protein Pept. Lett., 2004, 11, 331–337 CAS.
- J. Barre, J. M. Chamouard, G. Houin and J. P. Tillement, Equilibrium dialysis, ultrafiltration, and ultracentrifugation compared for determining the plasma–protein-binding characteristics of valproic acid, Clin. Chem., 1985, 31, 60–64 CAS.
- B. K. Bozoglan, S. Tunc and O. Duman, Investigation of neohesperidin dihydrochalcone binding to human serum albumin by spectroscopic methods, J. Lumin., 2014, 155, 198–204 CAS.
- G. F. Shen, T. T. Liu, Q. Wang, M. Jiang and J. H. Shi, Spectroscopic and molecular docking studies of binding interaction of gefitinib, lapatinib and sunitinib with bovine serum albumin (BSA), J. Photochem. Photobiol., B, 2015, 153, 380–390 CrossRef CAS PubMed.
- T. Peters, All About Albumin: Biochemistry, Genetics, and Medical Applications, Academic Press, San Diego, CA, 1996 Search PubMed.
- L. Painter, M. M. Harding and P. J. Beeby, Synthesis and interaction with human serum albumin of the first 3,18-disubstituted derivative of bilirubin, J. Chem. Soc., Perkin Trans. 1, 1998, 18, 3041–3044 Search PubMed.
- Y. H. Chen, J. T. Yang and H. M. Martinez, Determination of the secondary structures of proteins by circular dichroism and optical rotatory dispersion, Biochemistry, 1972, 11, 4120–4131 CAS.
- S. R. Feroz, S. B. Mohamad, N. Bujang, S. N. A. Malek and S. Tayyab, Multispectroscopic and molecular modeling approach to investigate the interaction of flavokawain B with human serum albumin, J. Agric. Food Chem., 2012, 60, 5899–5908 CAS.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, New York, 3rd edn, 2006 Search PubMed.
- O. K. Abou-Zied and O. I. Al-Shihi, Characterization of subdomain IIA binding site of human serum albumin in its native, unfolded, and refolded states using small molecular probes, J. Am. Chem. Soc., 2008, 130, 10793–10801 CAS.
- S. Bi, L. Ding, Y. Tian, D. Song, X. Zhou, X. Liu and H. Zhang, Investigation of the interaction between flavonoids and human serum albumin, J. Mol. Struct., 2004, 703, 37–45 CAS.
- G. Sancataldo, V. Vetri, V. Fodera, G. D. Cara, V. Militello and M. Leone, Oxidation enhances human serum albumin thermal stability and changes the routes of amyloid fibril formation, PLoS One, 2014, 9, e84552 Search PubMed.
- D. S. Goodsell, G. M. Morris and A. J. Olson, Automated docking of flexible ligands: applications of AutoDock, J. Mol. Recognit., 1996, 9, 1–5 CAS.
- M. F. Sanner, Python: A programming language for software integration and development, J. Mol. Graphics Modell., 1999, 17, 57–61 Search PubMed.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Cough, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, UCSF Chimera-a visualization system for exploratory research and analysis, J. Comput. Chem., 2004, 25, 1605–1612 CAS.
- J. A. Molina-Bolívar, F. Galisteo-González, C. C. Ruiz, M. M. Donnell and A. Parra, Spectroscopic investigation on the interaction of maslinic acid with bovine serum albumin, J. Lumin., 2014, 156, 141–149 Search PubMed.
- E. Guercia, C. Forzato, L. Navarini and F. Berti, Interaction of coffee compounds with serum albumins. Part II: Diterpenes, Food Chem., 2016, 199, 502–508 CrossRef CAS PubMed.
- G. Wang, D. Wang, X. Li and Y. Lu, Exploring the binding mechanism of dihydropyrimidinones to human serum albumin: Spectroscopic and molecular modeling techniques, Colloids Surf., B, 2011, 84, 272–279 CAS.
- J. Wang, C. Xiang, F. F. Tian, Z. Q. Xu, F. L. Jiang and Y. Liu, Investigating the interactions of a novel anticancer delocalized lipophilic cation and its precursor compound with human serum albumin, RSC Adv., 2014, 4, 18205–18216 CAS.
- A. Sulkowska, Interaction of drugs with bovine and human serum albumin, J. Mol. Struct., 2002, 614, 227–232 CrossRef CAS.
- Y. V. Il'ichev, J. L. Perry and J. D. Simon, Interaction of ochratoxin A with human serum albumin. A common binding site of ochratoxin A and warfarin in subdomain IIA, J. Phys. Chem. B, 2002, 106, 460–465 CrossRef.
- B. Tu, Y. Wang, R. Mi, Y. Ouyang and Y. J. Hu, Evaluation of the interaction between naringenin and human serum albumin: Insights from fluorescence spectroscopy, electrochemical measurement and molecular docking, Spectrochim. Acta, Part A, 2015, 149, 536–543 CAS.
- M. Saeidifar, H. Mansouri-Torshizi and A. A. Saboury, Biophysical study on the interaction between two palladium(II) complexes and human serum albumin by multispectroscopic methods, J. Lumin., 2015, 167, 391–398 CrossRef CAS.
- L. Trnková, I. Boušová, V. Staňková and J. Dršata, Study on the interaction of catechins with human serum albumin using spectroscopic and electrophoretic techniques, J. Mol. Struct., 2011, 985, 243–250 CrossRef.
- S. Tunc, O. Duman, I. Soylu and B. K. Bozoglan, Spectroscopic investigation of the interactions of carbofuran and amitrol herbicides with human serum albumin, J. Lumin., 2014, 151, 22–28 CrossRef CAS.
- W. R. Ware, Oxygen quenching of fluorescence in solution: an experimental study of the diffusion process, J. Phys.
Chem., 1962, 66, 455–458 CAS.
- S. Tunc, A. Çetinkaya and O. Duman, Spectroscopic investigations of the interactions of tramadol hydrochloride and 5-azacytidine drugs with human serum albumin and human hemoglobin proteins, J. Photochem. Photobiol., B, 2013, 120, 59–65 CAS.
- Q. Wang, Q. Sun, P. Tang, B. Tang, J. He, X. Ma and H. Li, Determination of potential main sites of apixaban binding in human serum albumin by combined spectroscopic and docking investigations, RSC Adv., 2015, 5, 81696–81706 CAS.
- T. S. G. Olsson, M. A. Williams, W. R. Pitt and J. E. Ladbury, The thermodynamics of protein–ligand interaction and solvation: Insights for ligand design, J. Mol. Biol., 2008, 384, 1002–1017 CrossRef CAS PubMed.
- I. M. Klotz, Physicochemical aspects of drug–protein interactions: A general perspective, Ann. N. Y. Acad. Sci., 1973, 226, 18–35 CAS.
- P. D. Ross and S. Subramanian, Thermodynamics of protein association reactions: Forces contributing to stability, Biochemistry, 1981, 20, 3096–3102 CrossRef CAS PubMed.
- B. X. Huang, H. Y. Kim and C. Dass, Probing three-dimensional structure of bovine serum albumin by chemical cross-linking and mass spectrometry, J. Am. Soc. Mass Spectrom., 2004, 15, 1237–1247 CrossRef CAS PubMed.
- Y. Z. Zhang, J. Dai, X. P. Zhang, X. Yang and Y. Liu, Studies of the interaction between sudan I and bovine serum albumin by spectroscopic methods, J. Mol. Struct., 2008, 888, 152–159 CrossRef CAS.
- S. N. Khan, B. Islam, R. Yennamalli, A. Sultan, N. Subbarao and A. U. Khan, Interaction of mitoxantrone with human serum albumin: Spectroscopic and molecular modeling studies, Eur. J. Pharm. Sci., 2008, 35, 371–382 CrossRef CAS PubMed.
- M. S. Celej, G. G. Montich and G. D. Fidelio, Protein stability induced by ligand binding correlates with changes in protein flexibility, Protein Sci., 2003, 12, 1496–1506 CrossRef PubMed.
- A. Shrake and P. D. Ross, Biphasic denaturation of human albumin due to ligand redistribution during unfolding, J. Biol. Chem., 1988, 263, 15392–15399 CAS.
- D. Ajloo, H. Behnam, A. A. Saboury, F. Mohamadi-Zonoz, B. Ranjbar, A. A. Moosavi-Movahedi, Z. Hasani, K. Alizadeh, M. Gharanfoli and M. Amani, Thermodynamic and structural studies on the human serum albumin in the presence of a polyoxometalate, Bull. Korean Chem. Soc., 2007, 28, 730–736 CrossRef CAS.
- G. A. Pico, Thermodynamic features of the thermal unfolding of human serum albumin, Int. J. Biol. Macromol., 1997, 20, 63–73 CrossRef CAS PubMed.
- Z. Cheng, R. Liu and X. Jiang, Spectroscopic studies on the interaction between tetrandrine and two serum albumins by chemometrics methods, Spectrochim. Acta, Part A, 2013, 115, 92–105 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra12019a |
|
| This journal is © The Royal Society of Chemistry 2016 |