
Redox activity of lignite and its accelerating effects on the chemical reduction of azo dye by sulfide†
Juanjuan Lia,
Guangfei Liu*a,
Jiti Zhoua,
Aijie Wang*b,
Jing Wanga and
Ruofei Jina
aKey Laboratory of Industrial Ecology and Environmental Engineering, Ministry of Education, School of Environmental Science and Technology, Dalian University of Technology, Dalian 116024, China. E-mail: guangfeiliu@dlut.edu.cn
bState Key Laboratory of Urban Water Resource and Environment, Harbin Institute of Technology, Harbin 150090, China. E-mail: waj0578@hit.edu.cn
First published on 8th July 2016
Abstract
The search and development of an efficient and cost-effective redox mediator is essential for rapid decolorization of azo dye wastewater. Here, for the first time the electron shuttling activity of different lignite samples was assessed and utilized to promote azo dye reduction by sulfide. Mediated electrochemical reduction and oxidation analysis indicated that the lignite samples possessed a higher electron accepting capacity but negligible electron donating capacity. And the promotion effects of lignite samples seemed to be determined by their electron accepting capacities. It was found that the lignite-mediated decolorization performance increased with the increase of sulfide concentration (0–3.0 mM), lignite dosage (0–300 mg L−1) and salinity (0–6% NaCl). Over 80% decolorization could be kept in eight successive rounds of operation, revealing the persistent acceleration effects of lignite. Measurement and comparison of individual reaction rates not only further confirmed the redox mediator activity of lignite, but also identified the first step, i.e., reduction of lignite by sulfide to be the rate-limiting step of mediated azo dye decolorization. Additionally, redox transformation was observed with lignite's oxygenated moieties and iron components, which were believed to contribute to lignite's redox mediator activity. Based on the findings of this study, redox-active lignite could be used to enhance the treatment of wastewater containing azo dyes and other oxidative pollutants.
Introduction
Azo dyes are characterized by having one or more azo groups (–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–) linking aromatic rings in their molecules. They are widely used in food, printing, textile, and cosmetics, industries etc. because of their ease of production, cost-effectiveness, and stability.1,2 The loss of about 2–50% azo dyes during application makes azo dye wastewater a serious problem worldwide.3,4 The release of effluents containing azo dyes not only deteriorates the aesthetic appearance and transparency of water bodies, but also disturbs the aquatic ecosystems due to the toxicity and carcinogenicity of azo dyes and their degradation intermediates.5 Thus the treatment of azo dye wastewater has attracted much interest during the past few years. Generally, azo dyes are firstly reduced/decolorized under anaerobic or anoxic conditions to corresponding aromatic amines, some of which could then be further mineralized aerobically. Therefore sequential anaerobic–aerobic treatment has been widely accepted as the most effective strategy for complete removal of azo dye from wastewater.
N–) linking aromatic rings in their molecules. They are widely used in food, printing, textile, and cosmetics, industries etc. because of their ease of production, cost-effectiveness, and stability.1,2 The loss of about 2–50% azo dyes during application makes azo dye wastewater a serious problem worldwide.3,4 The release of effluents containing azo dyes not only deteriorates the aesthetic appearance and transparency of water bodies, but also disturbs the aquatic ecosystems due to the toxicity and carcinogenicity of azo dyes and their degradation intermediates.5 Thus the treatment of azo dye wastewater has attracted much interest during the past few years. Generally, azo dyes are firstly reduced/decolorized under anaerobic or anoxic conditions to corresponding aromatic amines, some of which could then be further mineralized aerobically. Therefore sequential anaerobic–aerobic treatment has been widely accepted as the most effective strategy for complete removal of azo dye from wastewater.
Azo dye reduction during wastewater treatment is usually realized through combination of biological and chemical processes. Besides the co-metabolic reduction of azo dye by microbial cells, azo dye decolorization by chemical reductants such as sulfide, cysteine, and dithionite, etc. that are generated from sulfate-reducing process or released by cell lysis cannot be neglected.6–9
The reduction of azo dye has been believed to be the rate-limiting step of dye-containing wastewater treatment. A lot of work has been conducted to speed up the reductive decolorization process. Most of these studies focused on the development and utilization of redox active substances, which could improve electron transfer between chemical reductants/microbial cells and azo dye.9–11 Soluble redox mediators including riboflavin, quinone compounds, and humic substances bearing abundant quinone groups have been shown to effectively promote chemical and biological azo dye reduction.12–14 Moreover, mediated reduction has also been proved effective for speeding up reductive transformation of many other contaminants such as perchlorate, nitroaromatics, U(VI), Cr(VI), nitrate, and polyhalogenated compounds, etc.15 However, these soluble redox mediators could be easily lost during discharge of treated wastewater and result in secondary pollution. Also, continuous dosing of soluble redox mediators to maintain stable reduction performance might not be acceptable during practical operation due to increased treatment cost. To solve these problems and develop more regenerable redox mediator, insoluble or solid-phase redox materials deserve further exploration. Initially, van der Zee et al. demonstrated that activated carbon possessing surface quinone groups could act as redox mediator to accelerate azo dye decolorization by anaerobic granular sludge and sulfide.16 In addition, other insoluble carbon materials were also used as redox mediators to promote the reductive transformation of contaminants. For example, graphene oxide and carbon nanotubes were found to be capable of facilitating chemical and microbial nitrobenzene reduction.17,18 More recently, our group found that reduced graphene oxide modified by anthraquinone compound could effectively catalyze azo dye reduction by sulfide and anaerobic sludge.19 Although satisfying acceleration effects were obtained, the costly and time-consuming preparation process and potential risk of engineered carbon (nano)materials limited their practical use. More attention still needs to be paid for continuous search and development of more environment-friendly and cost-effective redox mediators.
Lignite, also known as brown coal, is generally regarded as the lowest rank of coal due to its relatively poor heat content. The world's recoverable reserves of lignite are ca. 195 billion tonnes.20 It is mined all over the world and mainly used as a fuel for power generation. However, the conventional way of lignite combustion causes serious air pollution and decreasing social acceptance in China and many other developing countries. On the other hand, because it contains a large quantity of humic substances, lignite has been successfully used as fertilizer and soil conditioner to promote plant growth and yields. It could also act as sorbent of metal or organic pollutant to remediate contaminated aquifers, soils, and sediments.21 It has been found that both soluble and insoluble humic substances could promote chemical and biological reduction of oxidative pollutants.14,22 However, to the best of our knowledge, no information concerning lignite's activities of shuttling electron and accelerating pollutant reduction was available.
In the present study, the redox activity of lignite and its potential to improve abiotic reduction of azo dye were investigated. Lignite samples collected from different regions in China were characterized. After measurement of their electron donating and accepting capacities (EDC and EAC) with electrochemical method, the activities of different lignite samples for accelerating chemical reduction of azo dye in the presence of sulfide were compared. The effects of lignite dosage, sulfide concentration, salinity, and repeated uses, etc. on lignite-mediated dye decolorization were studied in detail. Moreover, the rate-limiting step and the potential accelerating mechanism of lignite-mediated reduction of azo dye were investigated.
Experimental
Chemicals and lignite samples
Sodium sulfide (Na2S·9H2O), azo dyes including acid red 27 (AR27), methyl orange (MO), metanil yellow G (MYG), reactive red X-3B (X-3B), reactive brilliant red K-2G (K-2G), and direct blue 71 (DB71) were purchased from Sinopharm (Fig. S1†). Diquat dibromide monohydrate (DQ, 99.5%) and 2,2′-azino-bis(3-ethylbenzthiazoline-6-sulfonic acid) diammonium salt (ABTS, >99%) were purchased from Sigma-Aldrich and were of chromatographic grade. All other chemicals were of analytical grade and used as received.Four different kinds of lignite samples (IML, SXL, YNL, and XJL) were obtained from Jinli coal mine of Inner Mongolia, Xinyuan coal mine of Shanxi, Zhaotong coal mine of Yunnan, and Dananhu coal mine of Xinjiang, respectively. Over 90% of proved lignite reserves in China were contributed by these four provinces.23,24 Besides, an anthracite sample (SXA) obtained from Jincheng coal mine of Shanxi was used as control. All coal samples were used after grinding and sieving by a 200 mesh sieve.
Characterization of lignite samples
The morphology and size of lignite samples were studied with scanning electron microscope (SEM, NOVA NanoSEM 450) and dynamic light scatter (DLS, Zetasizer Nano ZS90). The elementary composition of lignite samples was determined by elementary analyzer (Elementar Vario EL) and inductively coupled plasma spectroscopy (Perkinelmer Optima2000DV). The BET specific surface area of lignite samples was measured by surface area analyzer (Quantachrome NOVA4200e). The lignite samples were also characterized with Fourier transform infrared spectroscopy (FTIR, Bruker Equinox 55). The electronic binding energies of SXL under different redox states were measured by X-ray photoelectron spectrometer (XPS, Thermo Scientific K-Alpha). Fe(II) content in SXL was measured through HCl (37%) extraction and ferrozine assay.25,26 Total iron content in SXL was also determined with ferrozine assay after HCl extraction and hydroxylamine hydrochloride (10%) reduction.25,27 The amount of Fe(III) in SXL was calculated as the difference between its total iron and Fe(II) contents.EAC and EDC measurement
The redox states of different lignite samples were studied with mediated electrochemical reduction (MER) and oxidation (MEO) according to a previously reported method.28,29 The glass electrochemical cell was closed with a Teflon cover. Glass carbon electrode, Ag/AgCl electrode, and platinum plate electrode served as working electrode, reference electrode, and counter electrode, respectively. The counter electrode was separated from the working electrode with a cation exchange membrane. The solution used in electrochemical cell was 0.1 M phosphate buffer (80 mL, pH 7) containing 0.1 M potassium chloride. Chronoamperometry measurements were performed using an electrochemical workstation instrument (Chenhua CHI660D). When the electrode was equilibrated to desired potential values (Eh = −0.49 V for MER and +0.61 V for MEO), 1 mL of DQ stock solution (10 mM) for MER or 2 mL of ABTS stock solution (4.6 mM) for MEO were spiked, respectively. After re-attainment of constant background currents, 0.1 mL of lignite suspension (1 g L−1) was added into the cell. All aqueous solutions in electrochemical experiments were purged with nitrogen gas (99.9%) for 2 h before use. A reaction time of 2500 s was set to ensure the completion of the electrochemical reaction. The resulting current peaks were integrated to obtain the EAC and EDC values of specific lignite sample.Dye reduction experiments
Unless otherwise noted, the experiments were performed in serum bottles containing 50 mL phosphate buffer (50 mM), 0.3 mM azo dye, 3 mM sodium sulfide, and 300 mg L−1 lignite. The solution in serum bottle was initially purged with nitrogen gas (99.9%) for 30 min to remove oxygen. Then the bottles were transferred to an anaerobic glove box where sulfide and lignite were added. The sodium sulfide stock solution was freshly prepared each time. Serum bottles sealed with butyl rubber stoppers and Al crimp caps were then placed onto magnetic stirrers (900 rpm, 25 °C). At intervals, samples were taken from the experimental systems and filtered to remove lignite. The absorbance of the filtrate was measured with UV-vis spectrophotometer (Jasco V-560) at maximum wavelength of respective azo dyes (λmax = 520 nm for AR27, λmax = 465 nm for MO, λmax = 448 nm for MYG, λmax = 540 nm for X-3B, λmax = 510 nm for K-2G, and λmax = 582 nm for DB71).To investigate the effects of lignite dosage on azo dye decolorization, 0–300 mg L−1 lignite were applied in the decolorization system. To study the effects of reductant concentration, sulfide concentration in the bottles was ranged from 0.6 to 3.0 mM. To study the effects of salinity, 1–6% NaCl was added into the decolorization system.
To study the long-term stability and persistence of lignite-mediated azo dye reduction, repeated batch operations of AR27 decolorization were performed. When around 90% AR27 decolorization was achieved, the decolorization system was purged with N2 (99.9%) for 3 h to completely remove excess sulfide, which was confirmed by methylene blue colorimetric method.30 Then another 0.3 mM AR27 and 3.0 mM sulfide were added into the system to start next round decolorization. Eight rounds of lignite-mediated AR27 reduction were conducted in total. All experiments were performed at least three times.
Measurement of reaction rates
Lignite-mediated decolorization of azo dye could be divided into two steps, i.e., reduction of lignite by sulfide and azo dye decolorization by reduced lignite. To better understand the mediated process, the specific rates of SXL reduction by sulfide (v1) and AR27 reduction by reduced SXL (v2) were determined, respectively. The reduction of lignite by sulfide was performed in a serum bottle containing 50 mL phosphate buffer (50 mM), 3 mM sodium sulfide, and 300 mg L−1 lignite. The solution in serum bottle was initially purged with nitrogen gas (99.9%) for 30 min to remove oxygen. At an interval of 1 h, 1 mL of reduced SXL suspension was sampled, purged with nitrogen gas (99.9%) for 20 min (to remove unreacted sulfide), and then mixed with 100 μL of ferric citrate solution (25 mM). The concentration of generated Fe(II) was measured through ferrozine assay.26 On the other hand, when SXL was completely reduced (named SXLred thereafter), as indicated by no further increase of Fe(II) concentration, nitrogen gas (99.9%) was purged into the suspension for 2 h to removal excess sulfide. Then, 0.3 mM AR27 was added into the serum bottle to start AR27 reduction by SXLred.The rates of the two separate reduction reactions were calculated by following formulas:
| v1 = ne1(ct − c0)/(tcsul) | (1) |
| v2 = ne2(cAR0 − cARt)/(tcAR0) | (2) |
The direct reduction rates of AR27 by sulfide were investigated under sulfide-limited and AR27-limited conditions, respectively. Either 0.3 mM AR27 were reduced by 1.2–3.0 mM sulfide, or 0.3–1.2 mM AR27 were reduced by 3.0 mM sulfide.
Results and discussion
Characterization of lignite samples
SEM images of different coal samples were shown in Fig. 1. The lignite samples were aggregates of small lamellas or particles. Residual plant tissue structure (Fig. 1e) could still be observed in some lignite samples, which might suggest an incomplete coalification process. The lignite samples were also rich in micron-sized pores and gaps. In contrast, whole micrometer-scale particles possessing relatively smooth surface and very limited pore structures were observed in the anthracite control sample. The BET surface areas of lignite samples ranged from 3.0 to 12.3 m2 g−1 (Table 1). However, the BET surface area of SXA was not measurable, further indicating its low porosity. The surface area of these coal samples are much less than those of activated carbon and nanosized carbon materials, which have been reported to be capable of speeding up microbial reduction of azo dyes.31–33 | ||
| Fig. 1 SEM images of (a and b) SXL, (c) IML, (d) YNL, (e) XJL and (f) SXA. | ||
| Samples | Elemental analysis (% by weight) | BET surface area (m2 g−1) | |||||
|---|---|---|---|---|---|---|---|
| C | H | O | N | S | Fe | ||
| a NM, not measurable. | |||||||
| IML | 63.88 | 4.63 | 12.27 | 0.66 | 0.33 | 0.25 | 6.87 |
| SXL | 41.09 | 3.63 | 28.98 | 0.89 | 1.11 | 2.62 | 12.25 |
| YNL | 42.51 | 5.17 | 24.77 | 1.26 | 0.50 | 1.25 | 5.76 |
| XJL | 40.64 | 3.28 | 27.76 | 0.48 | 0.09 | 2.07 | 3.00 |
| SXA | 70.69 | 2.57 | 16.91 | 0.73 | 0.56 | 0.23 | NM |
As shown in Table 1, IML possessed much higher C content than the rest three lignite samples (SXL, YNL, and XJL), which generally had a C content of around 40%. And the highest C content (over 70%) was found with the anthracite sample (SXA). This is consistent with the fact that anthracite is the product of final coalification stage and coal of highest rank. The O contents of SXL, YNL, and XJL generally lay between 25% and 30%, whereas those of IML and anthracite were much lower. The N and S contents of most coal samples investigated here were lower than 1% except those of YNL and SXL. Fe was found in both lignite and anthracite samples. SXL had the highest Fe content, which was almost ten times higher than that of SXA.
All the lignite and anthracite samples were investigated with FTIR (Fig. 2). The peak intensities of –OH (3412 cm−1), CO (1610 cm−1) and C–O (1400 cm−1)34 of lignite samples studied here were much stronger than those of the anthracite control sample (SXA), indicating the presence of more oxygenated groups on the surface of lignite samples. The bands at 2852 and 2920 cm−1 could be attributed to aliphatic groups. And the peaks between 700 and 900 cm−1 were attributed to out-of-plane C–H bending vibrations in aromatic units.35 Moreover, the peak at 1038 cm−1 was ascribed to OSO group.36 And the weak OSO peak intensities of IML and XJL were basically consistent with their low sulfur contents (Table 1).
 | ||
| Fig. 2 FTIR spectra of different coal samples. | ||
The redox properties of lignite and anthracite samples were investigated using mediated electrochemical methods. To the best of our knowledge, no such study for coal sample has been conducted before. The reductive and oxidative current peaks of different lignite and anthracite samples in MER and MEO were all well-defined (Fig. 3). The number of electrons which were transferred to and from the coal samples was obtained through integration of the current peaks. As shown in Table 2, all the coal samples had much higher EAC values than their EDC ones, suggesting that most of the redox active groups in these tested samples were in their oxidized forms. Similar results were also found in previous studies of other redox active substances.28,29 The EAC values of lignite samples were 16–27 times higher than that of SXA, indicating that the tested lignite samples could accept much more electrons in comparison to SXA. However, no significant difference was found among EDC values of lignite and anthracite samples. The highest EAC value observed with SXL was even comparable to those of standard and reference fulvic acids and wood- and grass-derived biochar,28,29 which have been characterized as good redox mediator.
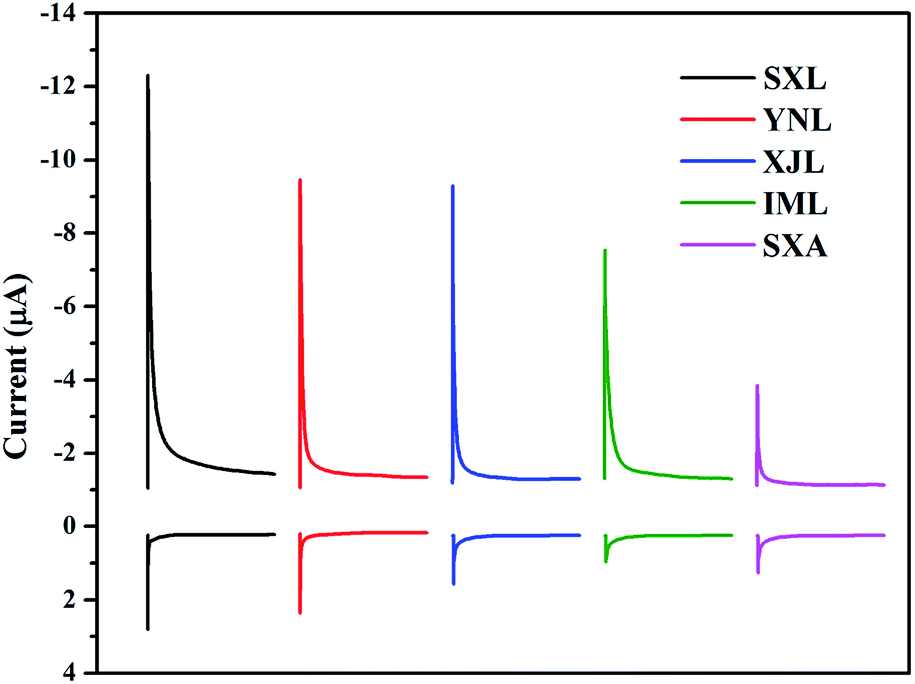 | ||
| Fig. 3 Reductive and oxidative current responses of five different coal samples. MER peaks were on the top of the figure and the other ones were MEO peaks. Reaction time for each coal sample was 2500 s. Current peaks were integrated to give transferred charge equivalents. | ||
| Samples | EAC (μmol e− per g) | EDC (μmol e− per g) |
|---|---|---|
| SXL | 514 ± 3 | 7.25 ± 0.5 |
| YNL | 427 ± 5 | 6.29 ± 1.0 |
| XJL | 410 ± 5 | 5.39 ± 0.8 |
| IML | 313 ± 5 | 1.60 ± 0.4 |
| SXA | 18.9 ± 2 | 3.08 ± 0.6 |
Promotion of chemical reduction of azo dye by lignite
The effects of different lignite and anthracite samples on sulfide-mediated reduction of azo dye were compared. No obvious removal of AR27 was found in control systems without the addition of sulfide (data not shown), which suggested the negligible AR27 adsorption capacity of different lignite samples and the important role of sulfide as reductant. This was also consistent with the EDC measurement results, i.e., the lignite samples themselves had very limited reduction capacity and could not reduce AR27 without exogenous reductant. As shown in Fig. 4, very limited reduction was obtained in system containing only AR27 and sulfide. Actually no reduction occurred in the initial 4 h and only around 7.2% AR27 was reduced in 9 h. The addition of different lignite samples all led to significant acceleration of AR27 reduction. The lignite-promoted decolorization process could be divided into two stages, a relatively slow initiation one followed by a faster decolorization stage. System added with SXL demonstrated the best mediated decolorization performance. Around 12.7% AR27 was reduced in 2 h with an initial decolorization rate of 12.7 mg L−1 h−1. Then the reduction rate increased to 21.3 mg L−1 h−1 and ultimately 95.3% AR27 disappeared in 9 h. For systems added with the rest three lignite samples (i.e., YNL, XJL, and IML), around 41.5%, 34.4%, and 13.3% AR27 were reduced in 6 h, with average reduction rates of 13.9 mg L−1 h−1, 11.6 mg L−1 h−1, and 4.5 mg L−1 h−1, respectively. After that, the average reduction rates were increased to 19.0 mg L−1 h−1, 17.6 mg L−1 h−1, and 6.9 mg L−1 h−1, respectively, and around 85.0%, 78.5%, and 31.0% AR27 were reduced in 9 h. The presence of anthracite control sample (SXA) could also improve AR27 decolorization by sulfide, although the promoting effects were poorer than those obtained with the addition of lignite samples. Around 23.7% AR27 was reduced in 9 h in the presence of 300 mg L−1 SXA. Notably, the rank of promoting capacities of different coal samples correlated well with that of their EAC values, suggesting that lignite could function as redox mediator and participate in electron transfer during chemical reduction of azo dye.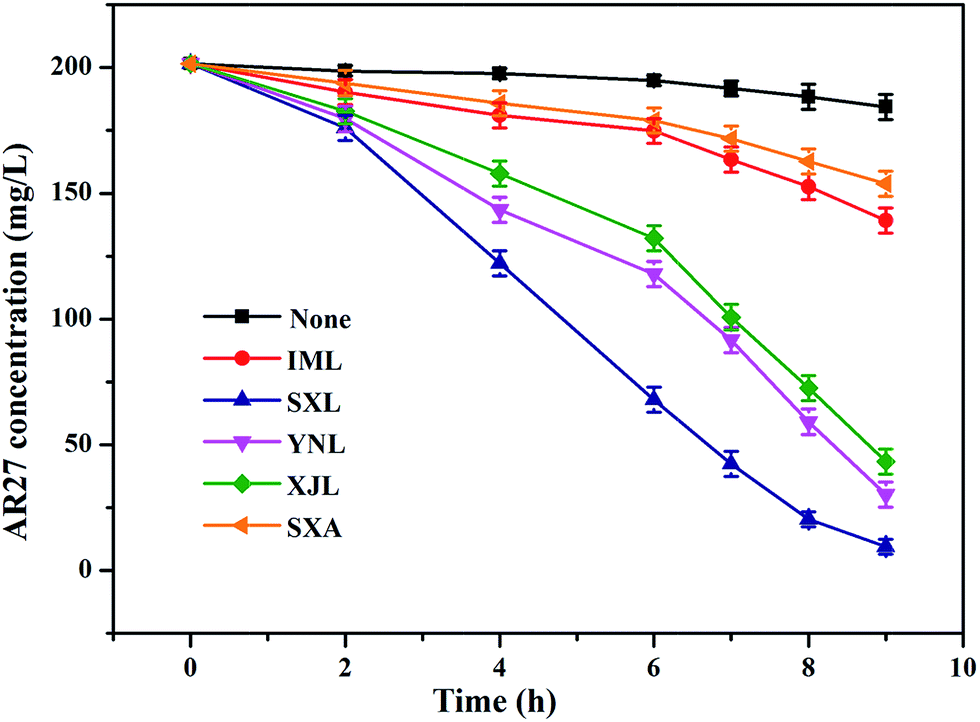 | ||
| Fig. 4 Effects of different coal samples on AR27 reduction by Na2S. Reaction conditions: 200 mg L−1 AR27, 3 mM Na2S, 300 mg L−1 coal samples and pH 7. | ||
SXL was then used to study the effects of lignite concentration on AR27 decolorization (Fig. 5a). Accelerating effects could be observed when SXL concentration was as low as 50 mg L−1. In 9 h, only 7.2% AR27 was reduced in the absence of lignite, whereas around 25.7%, 67.3%, and 86.0% AR27 decolorization were achieved with the addition of 50, 100, and 150 mg L−1 SXL, respectively. When the concentration of SXL was further increased to 200 and 300 mg L−1, over 90% decolorization could be obtained in 9 h. A linear relationship could be obtained when the average reduction rate in the initial 6 h was plotted against the SXL concentration in the reduction system (Fig. 5a, inset). This again indicated that the promotion effects of lignite increased with increase of added lignite concentration.
 | ||
| Fig. 5 Effect of (a) SXL concentration and (b) Na2S concentration on the reduction of AR27 mediated by SXL in Na2S-containing phosphate buffer. Reaction conditions: 200 mg L−1 AR27, pH 7, (a) 3 mM Na2S and 0–300 mg L−1 SXL, (b) 300 mg L−1 SXL and 0.6–3.0 mM Na2S. | ||
The effects of sulfide concentration on AR27 reduction in the presence of 300 mg L−1 SXL were also assessed (Fig. 5b). In the presence of 0.6 and 1.2 mM sulfide, around 35.4% and 64.9% AR27 were reduced in 9 h, respectively. When the sulfide concentration was further increased, although the overall decolorization efficiency continued to increase, the enhancement extent decreased gradually. Around 80.7%, 88.2%, and 94.5% AR27 were reduced in 9 h in the presence of 1.8, 2.4, and 3.0 mM sulfide, respectively. In a previous study assessing the promoting role played by activated carbon in azo dye decolorization, it was also demonstrated that an increase of sulfide concentration from 0.51 to 1.73 mM could significantly improve the decolorization performance.15 Moreover, a linear relationship was also found here between the average reduction rate and initial sulfide concentration (Fig. 5b, inset).
During textile dying process, large amounts of salts are generally used for dye fixation to fibers, organic contaminant separation (brine rinse) and dyestuff precipitation (salting out). Moreover, addition of NaOH into dye bath to increase pH could also lead to elevated sodium level in textile effluent. Therefore, salinity of dye wastewater must be considered during decolorization experiments. No significant difference in decolorization performance was found for direct reduction of AR27 by sulfide in the presence of 1–6% NaCl (data not shown). However, it was surprising to find that the presence of NaCl was conductive to lignite-mediated reduction of AR27. Better decolorization performance occurred under higher salinities (Fig. 6). Around 12.4% AR27 was reduced in the absence of NaCl in 2 h. Compared to that, around 16.6%, 22.7%, 29.2%, 33.1%, and 37.3% AR27 were reduced during the same period in the presence of 1%, 2%, 4%, 5%, and 6% NaCl, respectively. After 6 h, over 95% AR27 was decolorized when the NaCl concentration was higher than 2%. The initial reduction rate of AR27 increased linearly from 12.5 mg L−1 h−1 to 37.6 mg L−1 h−1 with the increase of NaCl concentration from 0 to 6% (Fig. 6, inset). As shown in Fig. S2,† the particle size of lignite in reduction system gradually decreased as time went on. The decreasing extent increased with the increase of salinity. Smaller average particle size was found with lignite particles in system having higher salinity. The presence of NaCl might speed up the removal of gypsum, soluble salts, and other matters adsorbed and coated on lignite.37–40 The accessible area and available redox sites of lignite could be increased with the decrease of its particle size. This might explain the better performance of lignite-mediated AR27 reduction under higher salinities.
 | ||
| Fig. 6 Effects of NaCl concentration on SXL-mediated reduction of AR27. Inset, relationship between initial reduction rate and NaCl concentration. Reaction conditions: 200 mg L−1 AR27, pH 7, 3 mM Na2S, and 300 mg L−1 SXL. | ||
The effects of lignite addition on chemical reduction of another five different azo dyes were also assayed. As shown in Fig. S3,† around 35.9% (in 2 h), 36.4% (in 3 h), 54.9% (in 14 h), 58.7% (in 48 h), and 62.5% (in 60 h) enhancement in decolorization efficiency were observed for reduction of MO, MYG, X-3B, K-2G, and DB71 in the presence of 300 mg L−1 SXL, respectively. Therefore the presence of lignite resulted in improved removal of all dyes investigated, suggesting the extensive effectiveness of lignite addition on accelerating azo dye reduction.
Repeated decolorization of 200 mg L−1 AR27 by sulfide in the presence of 300 mg L−1 SXL was studied. Over 80% decolorization could be kept in successive eight rounds of operation (Fig. S4†), which indicated that SXL had good stability and persistence in accelerating chemical reduction of azo dye. The decrease in decolorization efficiency from 93.9% of 1st run to 83.2% of 8th run might be due to the loss of SXL during repeated sampling and decolorization runs.
Determination of rate-limiting step
Results of electrochemical and decolorization studies suggested that lignite samples might function as redox mediators to accelerate electron transfer between sulfide and AR27. To further prove this, experiments were carried out to investigate the potential of SXL to accept electron from sulfide and reduction of AR27 by SXLred (Fig. S5†).For direct reduction of AR27 by sulfide, the reduction rates were determined to be 3.6 μmol e− per (h mM sulfide) (Fig. S5a†) and 32.5 μmol e− per (h mM dye) (Fig. S5b†) under sulfide-limited and AR27-limited conditions, respectively. On the other hand, the reduction rate of SXL by sulfide was calculated to be 17.4 μmol e− per (h mM sulfide) (Fig. S5c†), and the reduction rate of AR27 by SXLred was calculated to be 316.4 μmol e− per (h mM dye) (Fig. S5d†). By comparison, the rates of the two individual steps were about 4.8 and 9.7 times higher than that of direct AR27 reduction by sulfide. Therefore, it was quantitatively confirmed that SXL could act as redox mediator to accelerate AR27 reduction by sulfide. Moreover, the first step, i.e., the reduction of SXL by sulfide was identified as the rate-limiting step of SXL-mediated AR27 reduction by sulfide.
It should be noted that during its chemical reduction by sulfide, around 1.8 mmol electrons were transferred to each gram of SXL (calculated according to Fig. S5c†). This was 3.5 times higher than that determined by MER. This might be due to the difference in reducible groups between chemical and electrochemical methods.
Potential mechanism of lignite-mediated AR27 reduction
The C1s spectra of initial SXL (SXLini), SXLred, and SXLend (SXLred at the end of reaction with AR27) were shown in Fig. 7. The peak intensities of C–O bond at 286.1 eV and O–CO bond at 287.6 eV decreased after reaction with sulfide, but then went up to certain extent at the end of AR27 reduction. On the other hand, the peak intensity of C–C bond at 284.4 eV increased after reduction by sulfide, but then decreased at the end of AR27 reduction. The above findings suggested that SXL could act as redox mediator and participate in electron transfer from sulfide to AR27 through redox transformation of its oxygenated groups.
 | ||
| Fig. 7 C1s XPS spectra of (a) SXLini, (b) SXLred, and (c) SXLend. | ||
Iron minerals like hematite, ferrihydrite and magnetite have also been found capable of promoting the reduction of pollutants through redox cycle of Fe(III)/Fe(II).41–43 Zhang et al. found that iron fraction in a Fe–humic acid complex contributed to its electron-mediating activity.44 It was well-known that organic iron and different forms of Fe-bearing mineral phases such as pyrite, siderite, goethite and hematite existed in coals.45–47 Thus it is interesting to assay whether Fe components of lignite participated in mediated azo dye reduction. As shown in Fig. 8, the total Fe content in SXL was determined to be about 0.74 mmol g−1 SXL, among which 92% was found as Fe(III). During the two-step reaction, i.e., reduction of SXL by sulfide and reduction of AR27 by SXLred, the Fe(III) content in SXL sample firstly decreased from 0.68 mmol g−1 to 0.03 mmol g−1, and then went up again to 0.49 mmol g−1. Correspondingly, the Fe(II) content in SXL sample was observed to increase from 0.06 mmol g−1 to 0.72 mmol g−1 after being reduced by sulfide, and then returned to 0.24 mmol g−1 after incubation with AR27. Therefore, iron components in lignite might also be involved in accelerating azo dye reduction by sulfide.
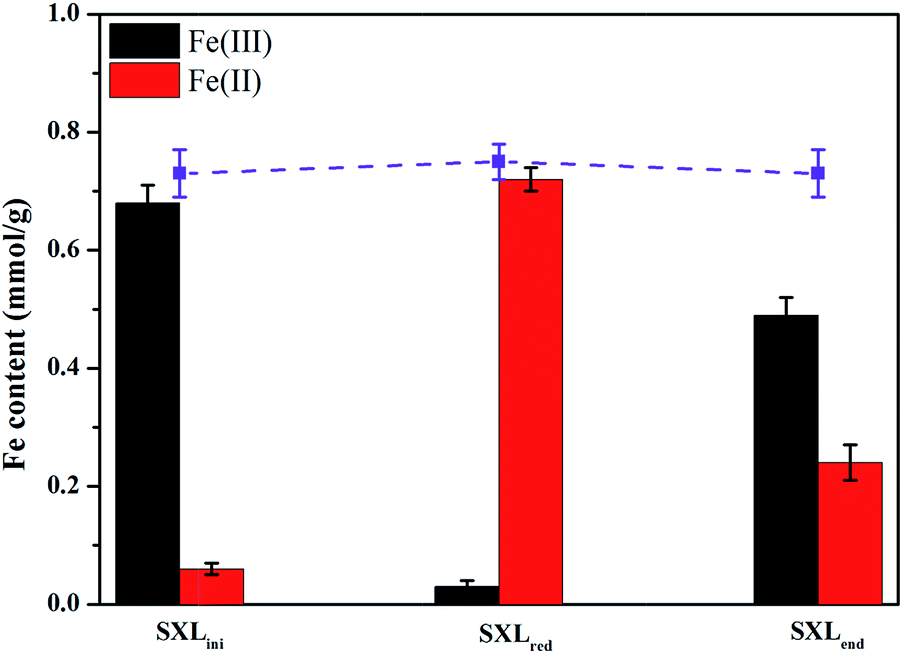 | ||
| Fig. 8 Variation of Fe(III)/Fe(II) contents in SXL of different redox states. The square symbol indicated the total Fe content in each sample. | ||
Conclusions
Lignite was for the first time found to be capable of acting as redox mediator to promote azo dye reduction by sulfide. Different lignite samples generally existed as aggregates of small lamelles/particles, possessed various oxygenated moieties on their surfaces, and demonstrated higher EAC values and limited EDC values during electrochemical analysis. Moreover, lignite samples having higher EAC values always showed better promoting effects. The increase of sulfide concentration, SXL dosage and salinity generally led to enhanced performance of mediated azo dye decolorization. And stable promotion effects were obtained with SXL in up to eight rounds of repeated uses. The reduction of lignite by sulfide was found to be the rate-limiting step of mediated azo dye decolorization. The oxygen-containing groups and iron components of the lignite were suggested to contribute to its electron shuttling activity and acceleration capacity. Considering the rich reserve and low cost of lignite, this lowest rank coal might be much more appropriate than most known redox mediators (quinones, humic substances, activated carbon, and carbon nanomaterials, etc.) for application in practical wastewater treatment, which will be tested in following studies.Acknowledgements
We thank the Natural Science Foundation of China (No. 51478076) and Open Project of State Key Laboratory of Urban Water Resource and Environment, Harbin Institute of Technology (No. QAK201530) for partial support of this study.References
- X. Meng, G. Liu, J. Zhou, Q. S. Fu and G. Wang, Bioresour. Technol., 2012, 114, 95–101 CrossRef CAS PubMed.
- S. Modi, B. Pathak and M. H. Fulekar, J. Environ. Nanotechnol., 2015, 4, 37–46 CAS.
- J. M. Rosa, A. M. F. Fileti, E. B. Tambourgi and J. C. C. Santana, J. Cleaner Prod., 2015, 90, 60–65 CrossRef CAS.
- T. R. Mota, C. G. Kato, R. A. Peralta, A. Bracht, G. R. de Morais, M. L. Baesso, C. G. M. de Souza and R. M. Peralta, Water, Air, Soil Pollut., 2015, 226, 1–11 CrossRef CAS.
- T. Chiong, S. Y. Lau, Z. H. Lek, B. Y. Koh and M. K. Danquah, J. Environ. Chem. Eng., 2016, 4, 2500–2509 CrossRef CAS.
- D. Prato-Garcia, F. J. Cervantes and G. Buitrón, J. Hazard. Mater., 2013, 250, 462–468 CrossRef PubMed.
- H. Li, Z. Xiong, X. Dai and Q. Zeng, Dyes Pigm., 2012, 94, 55–59 CrossRef CAS.
- L. Tan, Y. Liu, Q. Yang, X. Li, X. Wu, B. Gong, Y. Shen and Z. Shao, Chem. Commun., 2016, 52, 954–957 RSC.
- J. Sun, W. Li, Y. Li, Y. Hu and Y. Zhang, Bioresour. Technol., 2013, 142, 407–414 CrossRef CAS PubMed.
- B. Subash, B. Krishnakumar, A. J. F. N. Sobral, C. Surya, N. A. A. John, A. Senthilraja, M. Swaminathan and M. Shanthi, Sep. Purif. Technol., 2016, 164, 170–181 CrossRef CAS.
- M. Palencia, Á. A. Arrieta and M. Melendrez, Colloid Polym. Sci., 2015, 293, 3025–3032 CAS.
- L. R. Martins, B. E. L. Baêta, L. V. A. Gurgel, S. F. de Aquino and L. F. Gil, Ind. Crops Prod., 2015, 65, 454–462 CrossRef CAS.
- X. Meng, G. Liu, J. Zhou and Q. S. Fu, Bioresour. Technol., 2014, 151, 63–68 CrossRef CAS PubMed.
- C. M. Martínez, L. B. Celis and F. J. Cervantes, Appl. Microbiol. Biotechnol., 2013, 97, 9897–9905 CrossRef PubMed.
- F. P. van der Zee and F. J. Cervantes, Biotechnol. Adv., 2009, 27, 256–277 CrossRef CAS PubMed.
- F. P. van der Zee, I. A. Bisschops and G. Lettinga, Environ. Sci. Technol., 2003, 37, 402–408 CrossRef CAS PubMed.
- H. Fu and D. Zhu, Environ. Sci. Technol., 2013, 47, 4204–4210 CrossRef CAS PubMed.
- F. Yan, Y. He, C. Wu, Y. Cheng, W. Li and H. Yu, Environ. Sci. Technol. Lett., 2013, 1, 128–132 CrossRef.
- H. Lu, H. Zhang, J. Wang, J. Zhou and Y. Zhou, RSC Adv., 2014, 4, 47297–47303 RSC.
- V. R. Patel, D. S. Upadhyay and R. N. Patel, Energy, 2014, 78, 323–332 CrossRef CAS.
- M. Simmler, L. Ciadamidaro, R. Schulin, P. Madejón, R. Reiser, L. Clucas, P. Weber and B. Robinson, Environ. Sci. Technol., 2013, 47, 4497–4504 CrossRef CAS PubMed.
- F. J. Cervantes, C. M. Martínez, J. Gonzalez-Estrella, A. Márquez and S. Arriaga, Appl. Microbiol. Biotechnol., 2013, 97, 2671–2679 CrossRef CAS PubMed.
- Y. Xu, Y. Zhang, G. Zhang and Y. Guo, Energy Convers. Manage., 2016, 114, 11–19 CrossRef CAS.
- S. Tang, L. Wang, X. Feng, Z. Feng, R. Li, H. Fan and K. Li, Fuel, 2016, 180, 194–204 CrossRef CAS.
- P. Zhu, W. Zeng, X. Xu, L. Cheng, X. Jiang and Z. Shi, Int. J. Miner., Metall. Mater., 2014, 21, 317–324 CrossRef CAS.
- J. Im, J. Lee and F. E. Löfflera, J. Microbiol. Methods, 2013, 95, 366–367 CrossRef CAS PubMed.
- R. Akiyama, N. Kanno, Y. Suzuki and K. Yamamoto, Glass Technol., 2015, 56, 37–42 Search PubMed.
- L. Klüpfel, M. Keiluweit, M. Kleber and M. Sander, Environ. Sci. Technol., 2014, 48, 5601–5611 CrossRef PubMed.
- M. Aeschbacher, M. Sander and R. P. Schwarzenbach, Environ. Sci. Technol., 2009, 44, 87–93 CrossRef PubMed.
- L. Zhang, S. Li, M. Hong, Y. Xu, S. Wang, Y. Liu, Y. Qian and J. Zhao, Org. Biomol. Chem., 2014, 12, 5115–5125 CAS.
- G. Mezohegyi, F. P. van der Zee, J. Font, A. Fortuny and A. Fabregat, J. Environ. Manage., 2012, 102, 148–164 CrossRef CAS PubMed.
- L. Pereira, R. Pereira, M. F. R. Pereira and M. M. Alves, Biotechnol. Bioeng., 2016, 113, 1194–1202 CrossRef CAS PubMed.
- V. K. Gupta, R. Kumar, A. Nayak, T. A. Saleh and M. A. Barakat, Adv. Colloid Interface Sci., 2013, 193, 24–34 CrossRef PubMed.
- B. Dong, G. Liu, J. Zhou, A. Wang, J. Wang, R. Jin and H. Lv, RSC Adv., 2015, 5, 97798–97806 RSC.
- W. Zhang, S. Jiang, K. Wang, L. Wang, Y. Xu, Z. Wu, H. Shao, Y. Wang and M. Miao, Int. J. Coal Prep. Util., 2015, 35, 39–50 CrossRef CAS.
- L. Li, J. Liu, G. Chen, L. Xu, N. Mushtaq, L. R. Sidra, R. Wang and X. Fang, High Perform. Polym., 2016 DOI:10.1177/0954008315624955.
- S. Zikeli, M. Kastler and R. Jahn, Geoderma, 2005, 124, 253–265 CrossRef CAS.
- G. Machulla, S. Zikeli, M. Kastler and R. Jahn, J. Plant Nutr. Soil Sci., 2004, 167, 449–456 CrossRef CAS.
- D. C. W. Tsang and A. C. K. Yip, J. Soils Sediments, 2014, 14, 936–947 CrossRef CAS.
- F. S. Cannon, F. S. Cannon, N. R. Brown, T. M. Byrne, P. Hou, R. Parette, X. Gu, C. C. Cash, C. N. Delgado and S. Hong, US Pat., 9 095 840, 2015.
- F. Luan, W. Burgos, L. Xie and Q. Zhou, Environ. Sci. Technol., 2010, 44, 184–190 CrossRef CAS PubMed.
- Y. Xu, Y. He, X. Feng, L. Liang, J. Xu, P. C. Brookesa and J. Wu, Sci. Total Environ., 2014, 473, 215–223 CrossRef PubMed.
- M. Munoz, Z. M. de Pedro, J. A. Casas and J. J. Rodriguez, Appl. Catal., B, 2015, 176, 249–265 CrossRef.
- C. Zhang, D. Zhang, Z. Li, T. Akatsuka, S. Yang, D. Suzuki and A. Katayama, Environ. Sci. Technol., 2014, 48, 6318–6325 CrossRef CAS PubMed.
- M. A. Ahmed, N. I. Aljuraide and M. J. Bles, Energy Sources, Part A, 2016, 38, 1591–1597 CrossRef CAS.
- B. Dai, X. Wu, A. D. Girolamo, J. Cashion and L. Zhang, Fuel, 2015, 139, 733–745 CrossRef CAS.
- F. Zhang, D. Xua, Y. Wang, M. D. Argyle and M. Fan, Appl. Energy, 2015, 145, 295–305 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra11930a |
| This journal is © The Royal Society of Chemistry 2016 |