DOI:
10.1039/C6RA11908E
(Paper)
RSC Adv., 2016,
6, 67794-67804
Density functional theory study of the substituent effect on the structure, conformation and vibrational spectra in halosubstituted anilines†
Received
7th May 2016
, Accepted 10th July 2016
First published on 11th July 2016
Abstract
A comparative density functional theory (DFT) study exploring the structural and spectroscopic properties of the complete set of halosubstituted anilines with the halogens being F, Cl and Br was carried out. This study aims at understanding the effect of the type, number and positions of halogen substituents on the inversion barrier, geometrical properties and vibrational frequencies. The compounds were exclusively predicted to exist in the near-planar pyramidal form. While the order of stability is noted to be independent of the type of halogen substituents, the size of the inversion barrier is predicted to be sensitive to their number and positions, in accordance with former theoretical and experimental reports. The higher number of halogens leads to a more pronounced planarity character of the amino group. The deactivating nature of halogen atoms is believed to enhance the lone-pair electron delocalization in the order of bromo- > chloro- > fluoroanilines. An unusually strong electron delocalization nature is predicted to exist in the case of tetra- and penta-substituted haloanilines.
1. Introduction
Halogenated anilines are widely used in the manufacture of agricultural agents, dyes, drugs and related intermediates.1–10 Understanding the influence of the high electronegativity and electron-releasing properties of halogen substituents on the chemical nature of aromatic compounds, in particular the aniline family, has been the focus of a number of research reports, and for that purpose ab initio calculations and density functional theory (DFT) methods were found to be useful.11–24 Geometrical parameters in the ground and excited states,11 vibrational assignments and normal coordinate analyses,12–14 and thermochemical behavior15,16 of fluoro-substituted anilines were predicted with the help of various computational methods. Enthalpies of formation of fluoroanilines were estimated at the B3LYP/6-31G(d) and BP86/6-31+G(d) levels of theory15 with the G3MP2B3 composite approach.16 The order of stability of monofluoroanilines was predicted to be in the order of meta > ortho > para; in fair agreement with experimental results.15 m,m′-Difluoroaniline was shown to be thermochemically the most stable isomer of the six difluoroanilines, while o,o′,p- and o,m,m′,p-isomers were shown to be the most stable of the trifluoro- and tetrafluoroanilines, respectively.15
Moreover, the molecular geometries,17–19 enthalpies of formation,20 and detailed infrared and Raman spectra vibrational assignments17,18 were investigated for a series of chloroaniline systems on the basis of theoretical methods. o,o′,p-Trichloroaniline and o,m,m′,p-tetrachloroaniline, for instance, were predicted by DFT-B3LYP, ab initio MP2 and MP4(SDQ) methods to exist predominantly in a near-planar form.18 o,o′,p-Trichloroaniline structure and vibrational assignments were compared with the corresponding tribromoaniline.19 Additionally, the vibrational frequencies for p-fluoro-, p-chloro- and p-bromoanilines have been reported along with the calculated potential energy distribution (PED) values to assist in providing conclusive assignments of the experimental infrared and Raman spectra.21
Fewer theoretical studies, however, on bromoanilines have been found in literature.22,23 Structural aspects of meta and para mono-substituted bromoanilines have been predicted at the BP86/6-31+G* and B3LYP/6-31+G* levels of theory to be similar to those for the parent aniline molecule.22 The o-bromo-, o,o′-dibromo- and o,o′,p-tribromoaniline molecules were revealed by DFT method to be thermodynamically the most stable among the mono-, di-, and tribromoaniline series, respectively.22 Vibrational assignments of the fundamental modes for o,o′,p-tribromoaniline based on the observed and theoretical polarization properties and the crystal structure consistency have been reported.23
Except for the DFT study carried out by Altarawneh et al.24 in which the geometrical and thermochemical properties of chloro-substituted anilines were compared, no other reports have introduced comprehensive understanding of the influence of the identity, number and position of halogen substituents on the structural and spectroscopic properties. This has motivated us to explore the halogenation effect in a broader aspect using first-principle calculations, and we believe it will provide useful background for future investigation on halogenated anilines, especially those which have not been explored experimentally yet. The outcome of this work furthers in particular the understanding the pattern of behavior of the amino group inversion and molecular geometry in the context of the electron density variation as a result of different halogen substitution possibilities. The investigation has been extended to predict the effect on the vibrational frequencies and NMR constants as a result of various halogenation scenarios.
2. Methodology
Gaussian 09 package25 was used to carry out geometrical optimization and vibrational frequency calculations. All possible isomeric forms were optimized using density functional theory (DFT) method with the hybrid B3LYP (Becke-three-parameter, Lee–Yang–Parr) functional, which was found to be a reliable quantum chemical tool for aniline derivatives and provides good compromise between accuracy and computational cost.26,27 The 6-311++G(d,p) basis set that was shown to give quite satisfactory geometry results for small organic systems,28 including halogenated molecules,29,30 was implemented throughout this study. The adopted basis set has produced good agreement, especially in the pattern of change due to different substitution manners, with available experimental NH2 inversion barrier (Table S1†) and geometrical (Table S2†) data. Moreover, values and trend of changes in inversion barriers predicted for chlorinated anilines in this study were comparable with those reproduced with a more extended basis set, namely B3LYP/6-311+G(3df,2p), which was in turn found to give good agreement with experimental results.24 The adopted atom numbering system is given in Scheme 1. Normal coordinate analyses were carried out for the stable conformers of each molecule,31,32 and potential energy distributions (PEDs) for the vibrational modes were calculated using Veda program33 which routinely tests the internal coordinates according to the atom type and geometry to generate suitable combinations of symmetry coordinates. The PED values were computed using Veda by tracing the force constant matrix on the basis of mass-weighted Cartesian coordinates which can be linearly transformed to valence-force coordinates used to develop potential energy parameters associated with the molecular stretching, bending and torsion components.33 Vibrational assignments were made on the basis of calculated PED values and with the help of GaussView graphical animation.34 The Natural Bond Order analysis was performed using the NBO program35 infused in the Gaussian 09 package at the DFT-B3LYP/6-311++G(d,p) level of theory. NMR calculations to predict the proton and 13C chemical shifts were carried out using the Gauge-Independent Atomic Orbital (GIAO) method.36
 |
| | Scheme 1 Atom numbering of halo-substituted anilines (X = H, F, Cl or Br). | |
3. Results and discussion
3.1. Conformational analysis
The order of stability of the four possible conformations shown in Fig. 1 for halo-substituted anilines was explored (Tables 1–3), and the near-planar pyramidal structure (denoted as Pym I) in which the two N–H bonds are symmetrically towards one side of the benzene ring was predicted to be the most stable. The lone pair bears the right symmetry to overlap with the π* orbitals of the ring,37 providing further stability to the molecule. Experimental results for aniline,38,39 fluoroanilines40,41 and chloroanilines42,43 confirm that Pym I configuration maintains the optimum orientation of the lone pair on the nitrogen atom in haloanilines. The Pym I ↔ P interchange corresponds to the NH2 inversion in the gas phase which is an important stereochemical property. A few inversion barriers, namely for aniline and its mono-substituted derivatives, were determined experimentally.38,44–47 Table S1† lists these values and compares them with the corresponding computed ones from the density functional B3LYP and second-order Møller–Plesset (MP2) theories. While the B3LYP approach underestimates actual barrier heights,48,49 the MP2 method tends to overestimate them, in consistence with previous reports.27,50 However, when compared with the far-IR experimental data,45 the former method has been noticed to be more reliable at predicting the trend of halogenation effect on the barrier height. The o- and p-fluoroanilines maintain greater barriers than chloro and bromo counterparts, with the latter two being comparable to each other, which has been reproduced fairly well using the B3LYP/6-311++G(d,p) level. This study, therefore, has adopted this cost-efficient level to explore the effect of halogenation pattern on the inversion barrier properties for the other multi-substituted derivatives that the literature doesn't reveal experimental studies associated with.
 |
| | Fig. 1 Optimized structures for the possible configurations of 2,3,6-trichloroaniline as calculated at the B3LYP/6-311++G(d,p) level of theory. | |
Table 1 Calculated relative energies (kcal mol−1) of the possible conformers of fluoroanilines calculated at the DFT-B3LYP/6-311++G(d,p) level of theorya
| |
Pym I |
P |
Pym IIa |
Pym IIb |
PP |
| Pym I = near-planar pyramidal, P = planar, Pym II = staggered pyramidal, PP = perpendicular. |
| Aniline |
0.00 |
0.78 |
5.52 |
|
8.83 |
| o |
0.00 |
0.64 |
7.41 |
8.82 |
9.67 |
| m |
0.00 |
0.63 |
6.98 |
7.00 |
9.32 |
| p |
0.00 |
0.90 |
5.42 |
|
7.74 |
| o,m |
0.00 |
0.54 |
6.51 |
6.83 |
9.83 |
| o,p |
0.00 |
0.79 |
4.44 |
6.83 |
8.47 |
| o,m′ |
0.00 |
0.51 |
6.32 |
8.65 |
10.19 |
| o,o′ |
0.00 |
0.55 |
8.00 |
|
10.30 |
| m,p |
0.00 |
0.76 |
5.40 |
5.74 |
8.20 |
| m,m′ |
0.00 |
0.50 |
7.25 |
|
9.85 |
| o,m,p |
0.00 |
0.65 |
5.15 |
7.09 |
8.58 |
| o,m,m′ |
0.00 |
0.42 |
6.85 |
8.75 |
10.37 |
| o,m,o′ |
0.00 |
0.43 |
7.72 |
7.94 |
10.46 |
| o,p,m′ |
0.00 |
0.64 |
5.06 |
7.41 |
8.89 |
| o,p,o′ |
0.00 |
0.68 |
6.91 |
|
9.01 |
| m,p,m′ |
0.00 |
0.61 |
5.80 |
|
8.70 |
| o,m,p,m′ |
0.00 |
0.51 |
5.99 |
7.50 |
9.12 |
| o,m,m′,o′ |
0.00 |
0.34 |
8.48 |
|
10.72 |
| o,p,m′,o′ |
0.00 |
0.55 |
6.66 |
7.02 |
9.13 |
| o,m,p,m′,o′ |
0.00 |
0.46 |
7.00 |
|
9.40 |
Table 2 Calculated relative energies (kcal mol−1) of the possible conformers of chloroanilines calculated at the DFT-B3LYP/6-311++G(d,p) level of theory
| |
Pym I |
P |
Pym IIa |
Pym IIb |
PP |
| Aniline |
0.00 |
0.78 |
5.52 |
|
8.83 |
| o |
0.00 |
0.46 |
7.00 |
8.94 |
10.40 |
| m |
0.00 |
0.63 |
6.78 |
6.86 |
9.13 |
| p |
0.00 |
0.72 |
6.21 |
|
8.56 |
| o,m |
0.00 |
0.38 |
7.21 |
9.05 |
10.74 |
| o,p |
0.00 |
0.43 |
6.20 |
8.31 |
10.04 |
| o,m′ |
0.00 |
0.36 |
6.59 |
8.75 |
10.63 |
| o,o′ |
0.00 |
0.26 |
8.93 |
|
12.06 |
| m,p |
0.00 |
0.60 |
5.99 |
6.29 |
8.80 |
| m,m′ |
0.00 |
0.50 |
6.36 |
|
9.41 |
| o,m,p |
0.00 |
0.36 |
6.50 |
8.77 |
10.42 |
| o,m,m′ |
0.00 |
0.29 |
6.88 |
9.45 |
11.00 |
| o,m,o′ |
0.00 |
0.15 |
9.25 |
9.78 |
12.34 |
| o,p,m′ |
0.00 |
0.35 |
6.66 |
8.42 |
10.21 |
| o,p,o′ |
0.00 |
0.25 |
9.34 |
|
11.61 |
| m,p,m′ |
0.00 |
0.49 |
6.10 |
|
9.00 |
| o,m,p,m′ |
0.00 |
0.31 |
7.27 |
9.02 |
10.66 |
| o,m,m′,o′ |
0.00 |
0.15 |
10.29 |
|
12.73 |
| o,p,m′,o′ |
0.00 |
0.20 |
9.09 |
9.73 |
11.96 |
| o,m,p,m′,o′ |
0.00 |
0.15 |
10.09 |
|
12.46 |
Table 3 Calculated relative energies (kcal mol−1) of the possible conformers of bromoanilines calculated at the DFT-B3LYP/6-311++G(d,p) level of theory
| |
Pym I |
P |
Pym IIa |
Pym IIb |
PP |
| Aniline |
0.00 |
0.78 |
5.52 |
|
8.83 |
| o |
0.00 |
0.45 |
7.05 |
9.27 |
10.43 |
| m |
0.00 |
0.65 |
7.38 |
7.50 |
9.09 |
| p |
0.00 |
0.69 |
7.14 |
|
8.73 |
| o,m |
0.00 |
0.39 |
7.41 |
9.15 |
10.82 |
| o,p |
0.00 |
0.40 |
7.01 |
8.42 |
10.23 |
| o,m′ |
0.00 |
0.32 |
6.78 |
8.92 |
10.58 |
| o,o′ |
0.00 |
0.24 |
9.75 |
|
12.15 |
| m,p |
0.00 |
0.62 |
6.45 |
6.16 |
8.95 |
| m,m′ |
0.00 |
0.49 |
6.77 |
|
9.29 |
| o,m,p |
0.00 |
0.33 |
6.49 |
9.25 |
10.76 |
| o,m,m′ |
0.00 |
0.30 |
6.92 |
9.45 |
11.07 |
| o,m,o′ |
0.00 |
0.16 |
9.35 |
10.02 |
12.53 |
| o,p,m′ |
0.00 |
0.36 |
6.47 |
8.70 |
10.39 |
| o,p,o′ |
0.00 |
0.21 |
9.52 |
|
11.88 |
| m,p,m′ |
0.00 |
0.49 |
6.18 |
|
9.07 |
| o,m,p,m′ |
0.00 |
0.27 |
7.54 |
9.20 |
10.88 |
| o,m,m′,o′ |
0.00 |
0.10 |
9.88 |
|
12.95 |
| o,p,m′,o′ |
0.00 |
0.16 |
10.20 |
9.39 |
12.37 |
| o,m,p,m′,o′ |
0.00 |
0.12 |
10.53 |
|
12.90 |
In addition, Tables 1–3 show that the order of stability is Pym I > planar > Pym II > PP, regardless of the type, number and position of the halogen substituents. For the staggered Pym II forms, although the N–H bonds are positioned in a way to minimize the hindrance with respect to the C–H or C–X bonds, the lone pair at nitrogen causes a great deal of destabilization up to 10 kcal mol−1 due to the steric interaction with halogens in the ortho positions. Comparing the relative stabilities of the Pym IIa and Pym IIb forms shows that when the electron lone pair is towards the halogen substituent side, 2–3 kcal mol−1 more destabilization occurs irrespective of the type of the halogen atoms.
Inspecting the calculated inversion barriers in anilines being studied reveals that they exhibit relatively low values (1–2 kcal mol−1). Not only the ring aromaticity facilitates the electron lone-pair inversion, but also the halogen substituents enhance the planarity of the NH2 group. Fig. 2 shows that the number, type and position of the halogen affect the size of the barrier. The higher the number of halogen substituents and the closer their position to the amino group, the more reduced the barrier size. Apparently, the barrier is sensitive to the increasing interaction of the halogen substituents with the amino moiety.
 |
| | Fig. 2 Calculated NH2 inversion barrier for aniline, fluoroanilines, chloroanilines and bromoanilines. | |
While the drop in the size of the inversion barrier in fluoroanilines is up to 50% of that in the parent aniline molecule, it could reach up to 80% drop in the case of chloro- and bromoanilines. This is probably due to the larger size of chlorine and bromine leading to more significant interaction with the amino group. The effect of the fluorine substituent in the para position in maintaining a relatively high inversion barrier is noteworthy compared to the rest of positions.
3.2. Geometrical parameters
Selected optimized bond lengths and angles of halosubstituted anilines as well as for aniline are compared in Fig. 3 and 4, as well as Fig. S1.† Experimental geometrical parameters for aniline and a number of its halo derivatives have been previously reported.39,41,42,51–54 An overall glance at the bond lengths of pluri-substitutions of aniline shows that halogen substituents do not significantly influence the interatomic distances; which is confirmed when the experimental bond distances of aniline derivatives listed in Table S2† are compared. For instance, the optimized C–N bond lengths of haloanilines presented in Fig. 3a indicate that halogen substituents cause a slight shortening (for most of the cases is not more than 0.05 Å which is comparable with experimental values) compared to aniline. The shortest C–N bond lengths calculated are those for tetra- and penta-chloro and bromoanilines, which indicates the effect of the higher number of bulky halogen substituents on the stiffness of the C–N bond. This is correlated well with the inversion barrier size (Fig. 2) as the slightly stiffer the C–N linkage is, the more feasible it is for the lone pair of electrons to flip over to the other side. From the average C–C bond lengths given in Fig. 3b, it can be seen that they increase in the case of the less electronegative halogen substituents (chlorine and bromine) but decrease in the case of the more electronegative substituent (fluorine). Reported experimental values in Table S2† for o-fluoro- and o-chloro- substituted derivatives are in line with that observation. The C–C bond distance is more perturbed when there is a higher number of a given halogen substituent (increase in the order of up to 0.1 Å).
 |
| | Fig. 3 Optimized (a) N–C and (b) (C–C)ave bond length (Å) for aniline, fluoroanilines, chloroanilines and bromoanilines. | |
 |
| | Fig. 4 Optimized (a) HNH (b) H1NC and (c) H2NC bond angle (degree) for aniline, fluoroanilines, chloroanilines and bromoanilines. | |
Furthermore, the H–N–H bond angle seems to be sensitive towards the type, number and position of the halogen substituents (Fig. 4a). In general, ortho and meta positions were noted to be the most influential on the size of the angle. Experimental H–N–H angles of m-fluoro (115°), o-chloro (114°) and m-chloro (114°) compared to p-fluoro (112°) derivatives as well as aniline (111°) affirm the trend predicted from theory (Table S2†). The order of influence of the position on the increasing size of the H–N–H angle is: ortho ≈ meta > para. That pattern is comparable to the results obtained in a prior B3LYP study that employed a split-valence basis set with d- and f-type polarized functions for heavy atoms.55 For tetra- and penta-chloro- and bromoanilines, the H–N–H angle are predicted to be up to 5° greater compared to aniline. The halogen substituents increase the H–N–H bond angle since they facilitate the planarity character of the amino group as explained earlier (Tables 1–3). The H–N–C bond angle values for the haloanilines are given in Fig. 4b and c. Chloro- and bromoanilines tend to have slightly larger bond angles than the fluoroaniline counterparts, while halogenated derivatives in general exhibit larger angles compared to the parent molecule. The angle size is proportional to the number of substituents across the benzene ring as a consequence of the decrease in the inversion barrier, which is in accordance with the earlier reported computational study for chloroaniline isomers.24
3.3. Natural bonding orbital (NBO) analysis
Computed NBOs are useful in understanding the distribution of electron density in halosubstituted anilines.56,57 They provide insights on the stability and reactivity of the systems being studied. Fig. 5a depicts the natural population of the nitrogen atom p-orbital. An appreciable level of electron occupation of π-symmetry could be predicted, and the electron occupancy was revealed to be in the order: fluorine (1.82029 − 1.86023e) > chlorine (1.79183 − 1.85069e) > bromine (1.78359 − 1.84870e). The smaller occupancy value noticed for substituted anilines compared to the parent molecule is attributed to the stronger delocalized condition in haloaniline.58 The deactivating nature of halogen atoms59 enhances the π-electron delocalization into the ring. Its strength increases with increasing the size of halogen atoms, which explains the electron occupancy trend. In terms of the substitution position, para-substituted haloanilines were predicted to have a relatively higher occupancy which is understood from the less interaction of para substituents compared to the respective ortho and meta positions. It can be also seen that (Fig. 5a) the π-electron population on nitrogen decreases with increasing the number of substituents as a result of increased interaction with the amino group.
 |
| | Fig. 5 (a) Lone-pair of electrons' occupancy, and (b) valence electron configuration of the amino nitrogen in aniline, fluoroanilines, chloroanilines and bromoanilines. | |
Moreover, it is noticed that halogens do not cause much influence on the orbital hybridization of C1 atom (Table S3†). On the other hand, the nitrogen atom valence configuration in haloanilines can be seen to significantly differ from that in the parent molecule. The hybridization tendency of corresponding haloanilines towards the planar sp2 environment in the aniline group was predicted in the order of bromoanilines < chloroanilines < fluoroanilines (Fig. 5b). A higher number of substituents (especially chlorines and bromines) drives the NH2 to be much more closely of sp2 nature, and therefore a great deal of planarity shall be observed for tetra- and penta-chloro- and bromo-substituted anilines. As shown in Fig. 6, the nitrogen atom lone pair occupancy correlates linearly with the inversion barriers in the three sets of haloaniline molecules. Bromoaniline and chloroaniline occupancies were slightly more sensitive to the variation of the size of the inversion barrier.
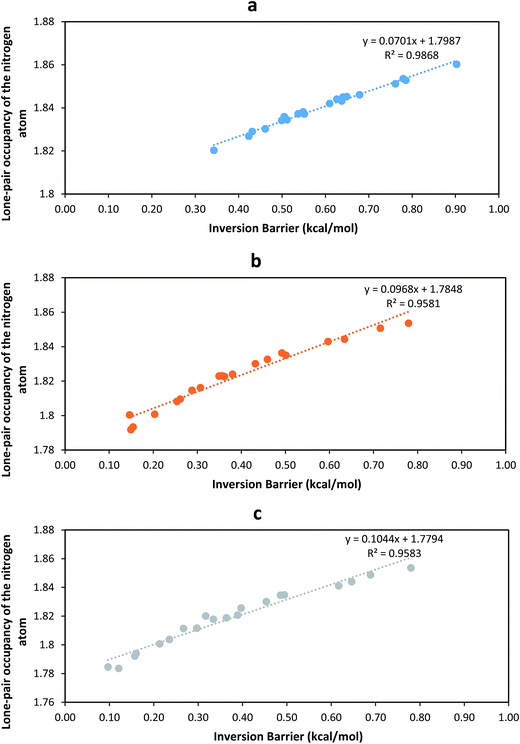 |
| | Fig. 6 Calculated lone-pair occupancy vs. the inversion barrier for (a) fluoroanilines, (b) chloroanilines and (c) bromoanilines. | |
3.4. Vibrational spectra
Vibrational frequencies associated with the ground-state Pym I configuration (Fig. 1) were computed, and reliable assignments have been made based on the calculated potential energy distributions (PEDs) as well as careful inspection of atom displacements. The computational method adopted was found to be in good agreement with previously reported studies of halosubstituted anilines.12–14,17–19,23 For instance, harmonic frequencies computed for p-haloaniline, monofluoroanilines and pentafluoroaniline, when scaled using appropriate scaling factors, were found comparable with the values predicted at the more extended B3LYP/6-311++G(df,pd) level as well as with the experimental ones.60–62 Haloanilines in the lowest-energy near-planar form undergo 36 molecular vibrations and, depending on the halogen substitution positions, can maintain Cs symmetry (vibrational modes span the irreducible representations: 20 A′ and 16 A′′) or C1 symmetry (all vibrations adopt A irreducible representations). Halogen substituents not only cause vibrational frequencies to shift but also affect the extent to which vibrational modes would couple with each other. Our analysis has focused on some key fundamentals to understand the effect of the type, position and number of halogen substituents on the vibrational spectra of haloanilines in the gas phase.
3.4.1. N–H stretching. The N–H stretching vibrations were predicted to exhibit slight shifts towards higher frequencies with ortho and meta substitutions and towards lower frequencies, but in less extent, for the para substitution in the infrared and Raman spectra of halogenated anilines. The N–H stretching frequency of fluoroanilines reported by Honda et al.63 using the jet-cooled technique showed a red-shift for o- and m-fluoro and a slight blue-shift for p-fluoroanilines supporting the results of Table 4 for mono-substituted fluoroanilines. The ortho substitution in particular causes a greater frequency change than other positions do. The shift to upward frequencies in the case of ortho can be explained in terms of the well-established intramolecular hydrogen bonding reported for fluoro64 and chloro56,65 derivatives. It is noticed from Table 4 (and Fig. S2†) that the vibrational shift becomes more apparent as the number of halogen substituents across the ring increases.62 While the asymmetric mode could shift by as much as 40 cm−1, the symmetric stretching vibration shift doesn't exceed 25 cm−1. Chloroanilines interestingly exhibit a relatively a stronger red shift as compared to fluoro and bromo counterparts.
Table 4 Calculated NH2 stretching vibrational frequencies (cm−1) and potential energy distributions (PEDs)a for aniline, fluoroanilines, chloroanilines and bromoanilines
| |
Fluoroanilines |
Chloroanilines |
Bromoanilines |
| Asym |
Sym |
Asym |
Sym |
Asym |
Sym |
| PED values as percentages are given in parentheses. |
| Aniline |
3666 (100) |
3569 (100) |
|
|
|
|
| o |
3678 (99) |
3576 (99) |
3682 (94) |
3577 (94) |
3679 (93) |
3572 (93) |
| m |
3674 (100) |
3575 (99) |
3674 (98) |
3575 (96) |
3674 (100) |
3575 (100) |
| p |
3662 (100) |
3566 (100) |
3669 (100) |
3571 (97) |
3670 (100) |
3572 (100) |
| o,m |
3683 (99) |
3581 (98) |
3689 (95) |
3583 (95) |
3686 (92) |
3577 (92) |
| o,p |
3672 (98) |
3573 (98) |
3684 (100) |
3579 (99) |
3683 (99) |
3576 (99) |
| o,m′ |
3685 (100) |
3582 (100) |
3688 (91) |
3582 (93) |
3686 (100) |
3577 (100) |
| o,o′ |
3688 (100) |
3582 (100) |
3696 (100) |
3587 (100) |
3689 (100) |
3578 (100) |
| m,p |
3670 (100) |
3573 (100) |
3675 (100) |
3576 (100) |
3676 (100) |
3577 (100) |
| m,m′ |
3683 (100) |
3582 (100) |
3682 (99) |
3581 (100) |
3681 (100) |
3581 (100) |
| o,m,p |
3677 (97) |
3576 (97) |
3689 (95) |
3583 (92) |
3686 (99) |
3577 (99) |
| o,m,m′ |
3691 (96) |
3586 (89) |
3694 (100) |
3587 (99) |
3691 (99) |
3580 (99) |
| o,m,o′ |
3691 (99) |
3584 (98) |
3701 (100) |
3590 (100) |
3692 (98) |
3580 (99) |
| o,p,m′ |
3679 (99) |
3578 (97) |
3688 (93) |
3582 (90) |
3686 (99) |
3578 (99) |
| o,p,o′ |
3679 (100) |
3575 (100) |
3696 (100) |
3587 (100) |
3691 (100) |
3579 (100) |
| m,p,m′ |
3677 (100) |
3578 (100) |
3680 (100) |
3580 (98) |
3680 (100) |
3580 (100) |
| o,m,p,m′ |
3684 (95) |
3581 (96) |
3694 (92) |
3587 (94) |
3691 (100) |
3581 (100) |
| o,m,m′,o′ |
3697 (98) |
3588 (99) |
3706 (100) |
3593 (100) |
3697 (100) |
3582 (100) |
| o,p,m′,o′ |
3685 (100) |
3580 (100) |
3701 (100) |
3590 (100) |
3694 (100) |
3580 (100) |
| o,m,p,m′,o′ |
3689 (99) |
3583 (96) |
3706 (100) |
3594 (98) |
3697 (100) |
3583 (100) |
3.4.2. C–N stretching. The C–N stretching vibration in aniline can be a useful indication of the magnitude of electron delocalization across the molecule. Unlike NH2 stretching, the C–N stretching vibration undergoes an appreciable degree of mixing with neighboring modes. Thus, we have made our assignments on the basis of the highest calculated PED values within the C–N stretching region (Table 5). While stiffness of the C–N bond steadily increases with the increase of the number of halogen substituents, a noticeable frequency shift (200 to 400 cm−1 upward) was predicted in the case of tetra- and penta-substitutions regardless the type of the halogen atom (Fig. S3-a†). Combining this observation with the aforementioned discussion pertaining the inversion barrier leads to the conclusion that a pronounced electron delocalization nature exists in the case of the tetra and penta isomers of aniline derivatives.
Table 5 Calculated C–N stretching vibrational frequencies (cm−1) and potential energy distributions (PEDs) for aniline, fluoroanilines, chloroanilines and bromoanilines
| |
Fluoroanilines |
Chloroanilines |
Bromoanilines |
| Aniline |
1277 (51) |
|
|
| o |
1330 (40) |
1287 (35) |
1286 (34) |
| m |
1335 (27) |
1344 (46) |
1288 (44) |
| p |
1295 (67) |
1300 (54) |
1300 (52) |
| o,m |
1360 (50) |
1336 (32) |
1334 (51) |
| o,p |
1328 (58) |
1323 (48) |
1323 (57) |
| o,m′ |
1331 (59) |
1288 (47) |
1285 (47) |
| o,o′ |
1316 (50) |
1309 (35) |
1309 (55) |
| m,p |
1346 (61) |
1316 (56) |
1312 (48) |
| m,m′ |
1373 (74) |
1302 (60) |
1294 (59) |
| o,m,p |
1368 (25) |
1331 (42) |
1325 (51) |
| o,m,m′ |
1400 (60) |
1306 (52) |
1298 (30) |
| o,m,o′ |
1680 (54) |
1601 (55) |
1590 (48) |
| o,p,m′ |
1345 (70) |
1298 (59) |
1292 (63) |
| o,p,o′ |
1542 (53) |
1312 (35) |
1311 (60) |
| m,p,m′ |
1405 (60) |
1309 (58) |
1297 (48) |
| o,m,p,m′ |
1433 (56) |
1460 (58) |
1439 (49) |
| o,m,m′,o′ |
1526 (59) |
1450 (51) |
1434 (56) |
| o,p,m′,o′ |
1522 (66) |
1474 (34) |
1303 (43) |
| o,m,p,m′,o′ |
1540 (30) |
1442 (55) |
1551 (57) |
3.4.3. Ring breathing. Ring breathing in benzene molecule reveals a characteristic peak in the gas-phase Raman spectrum at 992 cm−1.66 Upon benzene substitution, the position, intensity and degree of mixing of this mode are disturbed.18 The computed non-scaled ring-breathing frequency (830 cm−1) falls in good agreement with the experimental value67 (812 cm−1). The PED values (Table 6) indicate that in halosubstituted anilines the ring breathing is predominantly mixed with other vibrations, in particular with the C–N stretching and the H–C–C bending modes. In some extreme cases the ring bending mode dissolves out to the point it becomes spectroscopically insignificant for halosubstituted anilines. Generally, the significant low-side shift in frequency is associated with o,m-substitutions, while the high-side shift is associated with o,o′-substitutions (Fig. S3-b†). Para substations seem to have the least impact on the frequency value of the ring breathing mode especially for the three types of halogens. The shift in ring breathing frequencies for chloro- and bromoaniline isomers were noticed to closely follow a specific trend that is different from that of fluoroaniline counterparts. The variation in the ring breathing vibrational frequency, however, was noticed to be more sensitive towards the highly electronegative fluorine substituents.
Table 6 Calculated ring breathing vibrational frequencies (cm−1) and potential energy distributions (PEDs) for aniline, fluoroanilines, chloroanilines and bromoanilines
| |
Fluoroanilines |
Chloroanilines |
Bromoanilines |
| Aniline |
830 (31) |
|
|
| o |
1208 (21) |
1036 (28) |
1024 (49) |
| m |
1012 (35) |
1006 (58) |
1004 (53) |
| p |
858 (71) |
838 (44) |
835 (35) |
| o,m |
1073 (35) |
1039 (19) |
1023 (42) |
| o,p |
1072 (36) |
1052 (31) |
1037 (44) |
| o,m′ |
732 (24) |
716 (43) |
563 (56) |
| o,o′ |
1243 (29) |
1085 (47) |
1047 (51) |
| m,p |
969 (38) |
1032 (71) |
1017 (71) |
| m,m′ |
1028 (29) |
1004 (36) |
1000 (53) |
| o,m,p |
680 (61) |
775 (29) |
645 (41) |
| o,m,m′ |
1061 (16) |
958 (30) |
925 (37) |
| o,m,o′ |
460 (33) |
608 (35) |
741 (49) |
| o,p,m′ |
701 (49) |
739 (37) |
710 (47) |
| o,p,o′ |
811 (40) |
870 (48) |
1060 (52) |
| m,p,m′ |
1013 (30) |
1040 (42) |
1297 (48) |
| o,m,p,m′ |
1272 (28) |
1073 (32) |
1044 (37) |
| o,m,m′,o′ |
1101 (34) |
1099 (24) |
1054 (36) |
| o,p,m′,o′ |
1284 (33) |
1115 (21) |
1063 (42) |
| o,m,p,m′,o′ |
1311 (36) |
1119 (24) |
1065 (52) |
3.4.4. NH2 bending. The substituents effect on NH2 wagging, which is associated with the inversion motion of the amino group, and NH2 twisting vibrational frequencies in haloanilines can be revealed from Tables 7 and 8 (as well as Fig. S4†). These vibrations were observed in aniline68 at 541 cm−1 (574 cm−1 from DFT) and 277 cm−1 (282 cm−1). Calculated PED values (Tables 7 and 8) indicate that these bending modes are characteristic and maintain relatively large contribution to the assigned frequencies. These vibrations were predicted to be less significantly influenced by halogen substitutions in fluoroanilines compared to chloro- and bromo-analogs (Fig. S4†), indicating that the wagging and twisting vibrations of the amino group are more sensitive towards the size of the halogen substituents rather than the electronegativity. Halogen substituents lead to an increase in the planarity of the amino group19 and thus results with a more feasible wagging motion (Table 7). On the other hand, such an increasing planarity character somehow restricts the hydrogen twisting and is predicted to shift the respective frequencies to higher values (Fig. S4-b†). The NH2 bending vibrational frequencies are predicted to be more influenced by the closeness of the halogen substituents to the amino group as well as their larger size.
Table 7 Calculated NH2 wagging vibrational frequencies (cm−1) and potential energy distributions (PEDs) for aniline, fluoroanilines, chloroanilines and bromoanilines
| |
Fluoroanilines |
Chloroanilines |
Bromoanilines |
| Aniline |
574 (69) |
|
|
| o |
572 (50) |
509 (67) |
501 (73) |
| m |
528 (49) |
526 (42) |
524 (43) |
| p |
588 (75) |
561 (64) |
554 (64) |
| o,m |
527 (60) |
479 (66) |
467 (49) |
| o,p |
585 (34) |
569 (74) |
437 (59) |
| o,m′ |
523 (76) |
477 (71) |
465 (62) |
| o,o′ |
525 (68) |
437 (88) |
421 (89) |
| m,p |
557 (74) |
527 (78) |
519 (73) |
| m,m′ |
497 (80) |
497 (80) |
495 (74) |
| o,m,p |
553 (71) |
479 (80) |
467 (74) |
| o,m,m′ |
491 (75) |
447 (82) |
435 (44) |
| o,m,o′ |
511 (49) |
411 (50) |
390 (89) |
| o,p,m′ |
550 (74) |
479 (72) |
471 (66) |
| o,p,o′ |
549 (54) |
433 (78) |
413 (89) |
| m,p,m′ |
527 (79) |
508 (82) |
502 (82) |
| o,m,p,m′ |
523 (67) |
457 (51) |
438 (720) |
| o,m,m′,o′ |
462 (63) |
373 (86) |
364 (56) |
| o,p,m′,o′ |
529 (71) |
402 (49) |
383 (78) |
| o,m,p,m′,o′ |
503 (79) |
384 (54) |
354 (78) |
Table 8 Calculated NH2 twist vibrational frequencies (cm−1) and potential energy distributions (PEDs) for aniline, fluoroanilines, chloroanilines and bromoanilines
| |
Fluoroanilines |
Chloroanilines |
Bromoanilines |
| Aniline |
282 (98) |
|
|
| o |
331 (76) |
346 (93) |
346 (93) |
| m |
303 (88) |
305 (85) |
304 (65) |
| p |
255 (88) |
280 (96) |
285 (86) |
| o,m |
338 (66) |
362 (81) |
358 (82) |
| o,p |
293 (57) |
348 (74) |
351 (79) |
| o,m′ |
331 (59) |
360 (82) |
357 (84) |
| o,o′ |
357 (93) |
401 (91) |
409 (97) |
| m,p |
272 (90) |
291 (89) |
292 (85) |
| m,m′ |
309 (51) |
305 (96) |
303 (89) |
| o,m,p |
305 (64) |
355 (86) |
363 (75) |
| o,m,m′ |
381 (67) |
371 (72) |
370 (86) |
| o,m,o′ |
388 (71) |
419 (91) |
425 (64) |
| o,p,m′ |
309 (76) |
331 (73) |
362 (66) |
| o,p,o′ |
330 (73) |
396 (87) |
406 (96) |
| m,p,m′ |
284 (86) |
297 (93) |
298 (96) |
| o,m,p,m′ |
308 (52) |
373 (88) |
370 (74) |
| o,m,m′,o′ |
361 (75) |
434 (93) |
437 (85) |
| o,p,m′,o′ |
345 (76) |
411 (91) |
419 (91) |
| o,m,p,m′,o′ |
393 (95) |
427 (94) |
436 (94) |
3.5. 1H and 13C chemical shifts
The optimized structures were used to compute the 1H and 13C shielding constants (σ) for aniline, haloanilines and the reference TMS using the Gauge Independent Atomic Orbital (GIAO) method.34,69 A special attention was directed towards the 1H and 13C chemical shifts (δ) associated with the amino group hydrogens (H13 and H14) as well as C1, respectively (Fig. S5–S7†). In comparison with aniline, the C1 chemical shift of the haloanilines except for the m-substituted, m,m′-disubstituted haloanilines and the p-fluoroaniline were predicted to appear at relatively lower values. The carbon of the C–N bond in aniline is less shielded, therefore the up-field shift of carbon is probably due to the stronger C–N bond of the haloanilines which is proportional to the number of halo substituents. As carbon is viewed someway deshielded in the case of chloro- and bromo-substituted isomers regardless of the position and number of substituents, it appears to be more affected in the fluoro-substituted analogs, and in particular with the ortho position (Fig. S5†). The closeness of a highly electronegative group substituent causes the carbon atom to shift 20% up-field. On the other hand, it can be noticed (Fig. S6 and S7†) that the amino hydrogens are more affected by the interaction of the larger size Cl and Br substituents with the NH2 group leading to down-filed shifts (up to 35% in case of penta-halo) in the calculated 1H-NMR spectra. This could be considered an evidence of intramolecular N–H⋯Xortho bonding that is further enhanced by the presence of more halogen substituents in the benzene ring.70 From the NMR data, the intramolecular bonding increases with increasing the size of the halogen substituents in the order of fluoro < chloro ≈ bromo.
4. Conclusion
Density functional theory with the B3LYP/6-311++G(d,p) approach was employed to study the effect of F, Cl and Br substituents in the haloaniline series. In most of the cases, halogen substitutions are believed to result in a systematic influence on the conformational, structural and spectroscopic properties. This is due to the effect of the highly electronegative halogen atoms on the electronic nature of substituted anilines. While the number of substitutions has a direct impact on the barrier size of the NH2 inversion, the electronic occupancy was found sensitive towards both the number and the position of halogen substituents. These results have been found comparable with available experimental and theoretical reports. The outcome of this study helps in establishing a broad conformational- and spectroscopic-related understanding of the halosubstituted aniline family and shall provide useful insights for future experimental work.
References
- J. L. Radomski, Annu. Rev. Pharmacol., 1979, 19, 129–157 CrossRef CAS PubMed.
- R. D. Kimbrough, J. Environ. Sci. Health, Part B, 1980, 15, 977–992 CrossRef CAS PubMed.
- C. R. Worthing, The Pesticide Manual, The Lavenham Press, Lavenham Suffolk, 1987, vol. 8 Search PubMed.
- C. Tomlin, The Pesticide Manual, The Bath Press, Bath UK 10, 1995 Search PubMed.
- S. Leyva, V. Castanedo and E. Leyva, J. Fluorine Chem., 2003, 121, 171 CrossRef CAS.
- H. Cerecetto and W. Porcal, Mini-Rev. Med. Chem., 2005, 5, 57 CrossRef CAS PubMed.
- S. H. Gheewala and A. P. Annachhatre, Water Sci. Technol., 1997, 36, 5–63 CrossRef.
- P. C. Kearney and D. D. Kaufman, Degradation of herbicides, Marcel Dekker, Inc., New York, 1969 Search PubMed.
- The Merck Index – An Encyclopedia of Chemicals, Drugs and Biologicals, ed. M. J. O'Neil, Merck and Co., Inc., White House Station N. J, 2006, p. 230 Search PubMed.
- D. Ioffe and R. Frim, Kirk-Othmer Encyclopedia of Chemical Technology, John Wiley and Sons, New York, 1999–2013 Search PubMed.
- S. K. Pathak, R. Srivastava, A. K. Sachan, O. Prasad, L. Sinha, A. M. Asiri and M. Karabacak, Spectrochim. Acta, Part A, 2015, 135, 283–295 CrossRef CAS PubMed.
- I. Lopex-Tocon, M. Becucci, G. Pietraperzia, E. Castellucci and J. C. Otero, J. Mol. Struct., 2001, 565, 421 CrossRef.
- V. Mukherjee, K. Singh, N. P. Singha and R. A. Yadav, Spectrochim. Acta, Part A, 2009, 73, 44–53 CrossRef CAS PubMed.
- V. Mukherjee, N. P. Singh and R. A. Yadav, Spectrochim. Acta, Part A, 2009, 73, 249–256 CrossRef CAS PubMed.
- A. V. Manuel, et al., J. Phys. Chem. B, 2007, 111, 2052–2061 CrossRef PubMed.
- L. A. Curtiss, K. Raghavachari and J. A. Pople, J. Chem. Phys., 1999, 110, 7650 CrossRef.
- M. Govindarajan, M. Karabacak, S. Periandy and D. Tanuja, Spectrochim. Acta, Part A, 2012, 97, 231–245 CrossRef CAS PubMed.
- H. M. Badawi, W. Forner and A. A. Al-Saadi, J. Mol. Struct., 2009, 938, 41–47 CrossRef CAS.
- J. A. Faniran, H. F. Shurvell, D. A. Raeside, B. U. Petelenz and J. Korppi-Tommola, Can. J. Spectrosc., 1979, 24, 148–153 CAS.
- A. K. Rai, S. Kumar and A. Rai, Vib. Spectrosc., 2006, 42, 397–402 CrossRef CAS.
- P. M. Wojciechowski and D. Michalska, Spectrochim. Acta, Part A, 2007, 68, 948–955 CrossRef PubMed.
- M. A. V. Ribeiro da Silva, A. I. M. C. L. Ferreira and J. R. B. Gomes, Bull. Chem. Soc. Jpn., 2006, 79, 1852–1859 CrossRef CAS.
- R. M. P. Jaiswal, J. E. Katon and G. N. R. Tripathi, J. Chem. Phys., 1983, 79, 1–8 CrossRef CAS.
- I. Altarwneh, K. Altarawneh, A. H. Al-Muhtaseb, S. Alrawadieh and M. Altarawneh, Comput. Theor. Chem., 2012, 985, 30–35 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2013 Search PubMed.
- S. F. Sousa, P. A. Fernandes and M. J. Ramos, J. Phys. Chem. A, 2007, 111, 10439–10452 CrossRef CAS PubMed.
- K. C. Gross and P. G. Seybold, Int. J. Quantum Chem., 2000, 80, 1107–1115 CrossRef CAS.
- K. B. Wiberg, J. Comput. Chem., 2004, 11, 1342–1346 CrossRef PubMed.
- C. D. León, G. A. Echeverría, O. E. Piro, S. E. Ulic, J. L. Jios, M. Burgos Paci and G. A. Argüello, Chem. Phys., 2016, 472, 142–155 CrossRef.
- N. Sundaraganesan, S. Ilakiamani, H. Saleem, P. M. Wojciechowski and D. Michalska, Spectrochim. Acta, Part A, 2005, 61, 2995–3001 CrossRef CAS PubMed.
- W. Forner and H. M. Badawi, J. Mol. Model., 2001, 7, 288 CrossRef.
- E. B. Wilson, J. C. Decius and P. C. Cross, Molecular Vibrations, Mcgraw-Hill, York, 1995 Search PubMed.
-
(a) M. H. Jamróz, Vibrational Energy Distribution Analysis: VEDA 4, program, Warsaw, 2004–2010 Search PubMed;
(b) M. H. Jamróz, Spectrochim. Acta, Part A, 2013, 114, 220–230 CrossRef PubMed.
- R. Dennington II, T. Keith and J. Millam, GaussView, Version 5.0, Semichem Inc., Shawnee Mission, KS, 2009 Search PubMed.
- E. D. Glendening, J. K. Bandenhoop, A. E. Reed, J. E. Carpenter and F. Weinhood, NBO. 3.1 Program, Theoretical Chemistry Institute, University of Wisconsin, Madison, WI, 1996 Search PubMed.
- K. Wolinski, J. F. Hinton and P. Pulay, J. Am. Chem. Soc., 1990, 112, 8251–8260 CrossRef CAS.
- P. M. Wojciechowski, W. Zierkiewicz, D. Michalska and P. Hobza, J. Chem. Phys., 2003, 118, 1090 CrossRef.
- G. Roussy and A. Nonat, J. Mol. Spectrosc., 1986, 118, 180–188 CrossRef.
- D. Lister, J. Tyler, J. Høg and N. Larsen, J. Mol. Struct., 1974, 23, 253–264 CrossRef CAS.
- A. Nonat and G. Roussy, J. Mol. Spectrosc., 1985, 114, 13–22 CrossRef CAS.
- G. Cazzoli, D. Damiani and D. Lister, J. Chem. Soc., Faraday Trans. 2, 1973, 69, 119 RSC.
- A. Nonat, A. Bouchy and G. Roussy, J. Mol. Spectrosc., 1984, 108, 230–239 CrossRef CAS.
- A. Nonat, A. Bouchy and G. Roussy, J. Mol. Spectrosc., 1983, 99, 407–414 CrossRef CAS.
- J. Brand, D. Williams and T. Cook, J. Mol. Spectrosc., 1966, 20, 359–380 CrossRef CAS.
- M. Quack and M. Stockburger, J. Mol. Spectrosc., 1972, 43, 87–116 CrossRef CAS.
- N. Larsen, E. Hansen and F. Nicolaisen, Chem. Phys. Lett., 1976, 43, 584–586 CrossRef CAS.
- R. A. Kydd and P. J. Krueger, J. Chem. Phys., 1978, 69, 827–832 CrossRef CAS.
- S. Jeon, J. Choo, S. Kim, Y. Kwon, J. Kim, Y. Lee and H. Chung, J. Mol. Struct., 2002, 609, 159–167 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Acc. Chem. Res., 2008, 41(2), 157–167 CrossRef CAS PubMed.
- B. J. Lynch and D. G. Truhlar, J. Phys. Chem. A, 2001, 105, 2936–2941 CrossRef CAS.
- A. Hastie, D. G. Lister, R. L. McNeil and J. K. Tyler, J. Chem. Soc. D, 1970, 2, 108–109 RSC.
- S. N. Thakur, S. K. Tiwari and D. K. Rai, J. Mol. Struct., 1970, 5, 309 CrossRef CAS.
- A. Nonat, A. Bouchy and G. Roussy, J. Mol. Struct., 1983, 97, 83–86 CrossRef CAS.
- E. Csakvari and I. Hargittai, J. Phys. Chem., 1992, 96, 5837–5842 CrossRef CAS.
- A. Nonat and G. Roussy, J. Mol. Spectrosc., 1985, 114, 13–22 CrossRef CAS.
- S. Shaji and T. M. A. Rasheed, Spectrochim. Acta, Part A, 2001, 57, 337–347 CrossRef CAS.
- A. E. Reed, F. Weinhold and L. A. Curtiss, Chem. Rev., 1988, 88, 899–926 CrossRef CAS.
- A. K. Guha, C. Das and A. K. Phukan, J. Organomet. Chem., 2011, 696, 586–593 CrossRef.
- N. Aljamali, Int. J. Curr. Res. Chem. Pharm. Sci., 2014, 1, 121–151 Search PubMed.
- P. M. Wojciechowski and D. Michalska, Spectrochim. Acta, Part A, 2007, 68, 948–955 CrossRef PubMed.
- P. M. Wojciechowski, K. Helios and D. Michalska, Vib. Spectrosc., 2011, 57, 126–134 CAS.
- P. Wojciechowski, J. Fluorine Chem., 2013, 154, 7–15 CrossRef CAS.
- M. Honda, A. Fujii, E. Fujimaki, T. Ebata and N. Mikami, J. Phys. Chem. A, 2003, 107, 3678–3686 CrossRef CAS.
- D. Christen, D. Damiani and D. G. Lister, J. Mol. Struct., 1977, 41, 315–317 CrossRef CAS.
- V. K. Rai, S. B. Rai and D. K. Rai, Spectrochim. Acta, Part A, 2003, 59, 1299–1306 CrossRef.
- L. Goodman, A. Ozkabak and S. Thakur, J. Phys. Chem., 1991, 95, 9044–9058 CrossRef CAS.
- M. Castellá-Ventura and E. Kassab, Spectrochim. Acta, Part A, 1994, 50, 69–86 CrossRef.
- P. Wojciechowski, W. Zierkiewicz, D. Michalska and P. Hobza, J. Chem. Phys., 2003, 118, 10900 CrossRef CAS.
- R. Ditchfield, J. Chem. Phys., 1972, 56, 5688–5692 CrossRef CAS.
- M. E. Vaschetto, B. A. Retamal and A. P. Monkman, J. Mol. Struct.: THEOCHEM, 1999, 468, 209–221 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra11908e |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 






