
Preparation of a graphitic N-doped multi-walled carbon nanotube composite for lithium–sulfur batteries with long-life and high specific capacity†
 ab
ab
Abstract
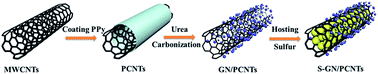
One of the challenges for lithium–sulfur batteries is a rapid capacity fading owing to the insulating of sulfur and Li2S2/Li2S compounds, the dissolving and consequent shuttling of polysulfide generated as intermediates during charge–discharge processes in the electrolyte. In this work, graphitic N-doped multi-wall carbon nanotube (GN/PNCNTs) composites are synthesized by in situ chemical polymerization and carbonization processes. The nitrogen doping in the GN/PNCNTs composite can effectively enhance chemisorption between sulfur and carbon, which can enable the uniform deposition of discharge products and lead to a high utilization and reversibility of active materials. Because of these technological superiorities, the as-prepared S-GN/PNCNTs cathode with a sulfur content of 60 wt% exhibits high initial specific capacity and excellent cycling stability at up to 600 cycles at 1C. Meanwhile, the rate capacities of the cathode are demonstrated from 0.5C to 6C with a specific capacity of 1178 mA h g−1 (the initial specific capacity) to 586 mA h g−1 (the 60th cycle).
 Please wait while we load your content...
Please wait while we load your content...

