
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Microwave-assisted synthesis of highly crystalline, multifunctional iron oxide nanocomposites for imaging applications†
Marc J. Williamsb,
Enrique Sáncheza,
Esther Rani Aluria,
Fraser J. Douglasc,
Donald A. MacLarenc,
Oonagh M. Collinsd,
Edmund J. Cussend,
James D. Budgee,
Lara C. Sanderse,
Martin Michaelise,
C. Mark Smalese,
Jindrich Cinatl Jrf,
Silvia Lorriogh,
Dirk Kruegerg,
Rafael T. M. de Rosales g and
Serena A. Corr*a
g and
Serena A. Corr*a
aSchool of Chemistry, University of Glasgow, Glasgow G12 8QQ, UK. E-mail: serena.corr@glasgow.ac.uk; Tel: +44 (0)141 3302274
bSchool of Physical Sciences, University of Kent, Canterbury CT2 7NH, UK
cSchool of Physics and Astronomy, University of Glasgow, Glasgow G12 8QQ, UK
dDepartment of Pure and Applied Chemistry, University of Strathclyde, Glasgow G1 1XL, UK
eCentre for Molecular Processing and School of Biosciences, University of Kent, Canterbury CT2 7NJ, UK
fInstitut für Medizinische Virologie, Klinikum der Goethe-Universität, Paul Ehrlich-Strasse 40, 60596 Frankfurt am Main, Germany
gDivision of Imaging Sciences and Biomedical Engineering, King’s College London, St Thomas’ Hospital, London, SE1 7EH, UK
hThe British Heart Foundation Centre of Excellence, Cardiovascular Division, King’s College London, London, UK
First published on 22nd August 2016
Abstract
We report a reproducible single-step, microwave-assisted approach for the preparation of multifunctional magnetic nanocomposites comprising superparamagnetic iron oxide (Fe3O4) cores, a polyelectrolyte stabilizer and an organic dye with no requirement for post-processing. The stabilisers poly(sodium 4-styrenesulfonate) (PSSS) and sodium polyphosphate (SPP) have been thoroughly investigated and from analysis using electron microscopy, dynamic light scattering measurements, magnetic hysteresis and magnetic resonance (MR) imaging, we show that the higher degree of Fe3O4 nanoparticle crystallinity achieved with the PSSS stabiliser leads to enhanced magnetic behaviour and thus better contrast agent relaxivity compared to the less crystalline, poorly defined particles obtained when SPP is employed as a stabiliser. We also demonstrate the potential for obtaining a multifunctional magnetic-fluorescent nanocomposite using our microwave-assisted synthesis. In this manner, we demonstrate the intimate link between synthetic methodology (microwave heating with a polyelectrolyte stabilizer) and the resulting properties (particle size, shape, and magnetism) and how this underpins the functionality of the resulting nanocomposites as agents for biomedical imaging.
1 Introduction
The choice of synthetic approach employed for the preparation of nanoparticles is of crucial importance when designing materials for a specific function. Of growing recent interest has been the development of routes to nanoparticles which afford great control over particle shape and composition, vital in our efforts to realise an intimate understanding of their unique properties. One example is the field of magnetic nanoparticles, a class of materials whose size-dependent magnetic properties opens up their potential applications for hyperthermic cancer therapy, site-specific drug delivery and contrast enhancement in magnetic resonance (MR) imaging.1–11 Control over the magnetic properties is desirable in order to tailor the candidate nanoparticle for a specific biomedical application.12 Magnetic nanoparticles may be synthesised in a variety of ways,13–15 including by co-precipitation,16,17 hydrothermal methods,18 and the decomposition of precursors at elevated temperatures.19,20 Aqueous routes, such as co-precipitation, are advantageous in that the particles are prepared in biologically tolerated solvents, but often lead to polydisperse nanoparticles which may display some loss of particle crystallinity.21 Advances made in the high temperature decomposition of organometallic precursors have led to highly crystalline, monodisperse nanoparticles, with a great degree of control over particle size.22 These routes, where organic solvents are employed, generate hydrophobic nanoparticles, with additional work-up required to transfer the particles to aqueous environments for subsequent biomedical use. A synthetic approach which produces nanoparticles with high crystallinity without the need for surface post-processing to induce hydrophilicity is therefore highly desirable.With these concerns in mind, we have employed microwave-assisted methods for the preparation of functionalised iron oxide nanoparticles, which are becoming increasingly attractive in materials chemistry. Microwave approaches to particle synthesis have led to dramatic decreases in reaction times and greater control over product formation.23–29 For example, the Niederberger group has developed microwave-assisted methods to prepare a range of nanoparticles of controlled sizes within minutes.30 Since the first reports of the microwave-promoted hydrothermal synthesis of sub-micron haematite powders,31 methods have been developed to prepare nanoparticles of uniform size distribution. Nanoparticles of haematite (α-Fe2O3) have been reported from the irradiation of hydrolysed iron salts,32 while more exotic α-Fe2O3 morphologies (cubes, rings and spindles) have been realised by Yu and co-workers by tailoring the reaction conditions to provide thermodynamic control over particle growth.33,34 Superparamagnetic maghemite (γ-Fe2O3) nanoparticles may be prepared through a microwave-treated co-precipitation reaction with a typical particle size of 10 nm obtained.35 Increases in nanoparticle crystallinity have been observed upon aging under microwave conditions, with post-processing using stearic acid resulting in stable organic ferrofluids.36 Uptake in endothelial cells of superparamagnetic citrate-coated Fe2O3 nanoparticles prepared by microwave-assisted methods have also recently been reported.37 Variation of reactant concentration in the microemulsion synthesis of Fe3O4 under microwave irradiation has resulted in monodisperse composites of variable sizes.38 Reaction time for the polyol synthesis of cobalt ferrite nanoparticles has been decreased to just one hour by combining microwave heating,39 while a variety of MFe2O4 ferrites (M = Zn, Ni, Mn, Co) have also been reported from microwave-hydrothermal methods.40
For their use as imaging agents, tailoring the surface chemistry of magnetic nanoparticles is essential.22,41–44 For example, Liong et al. have reported the preparation of a multifunctional nanocomposite for imaging and targeted drug delivery where iron oxide nanoparticles are phase transferred from an organic to an aqueous solution before silica coating and co-condensation of a fluorescent moiety render them multifunctional.45 Recently, Liu et al. have reported the use of polyacrylic acid in the synthesis of Fe3O4 nanoparticles to achieve tuneable particle cluster sizes of between 100 nm and 400 nm, which display good biocompatability.46 We have previously reported the use of polyelectrolytes for the in situ stabilisation of magnetic nanoparticles during a co-precipitation reaction, which has led to new developments in magnetic fluid preparation.47–49 Employing polyelectrolyte stabilisers in situ during nanoparticle nucleation and growth confers a high degree of stability induced by strong affinity of the anionic groups to the metal cations, with stable aqueous suspensions in the presence of a 0.5 T magnetic field obtained.
Here, we report the reproducible, repeatable preparation of bare Fe3O4 nanoparticles and the first use of microwave synthesis to obtain multi-coordinating polyelectrolyte-stabilised Fe3O4 nanoparticles and polyelectrolyte-stabilised magnetic fluids functionalised with a fluorescent dye according to Fig. 1. We are particularly interested in what effect, if any, the choice of polyelectrolyte stabiliser has on the resulting nanoparticle shape, size and crystallinity. The polyelectrolytes used were poly(sodium 4-styrenesulfonate) (PSSS), which has been employed previously as a stabiliser in a traditional co-precipitation reaction,48 and sodium polyphosphate (SPP) which was employed as an in situ stabiliser for the first time, where the sulfonate and phosphate groups, respectively, bind to the iron ions in solution prior to particle precipitation. In this manner, the polyelectrolytes act as stabilisers preventing the further growth of the nanoparticles, while at the same time promoting colloidal stability of the particles in water.
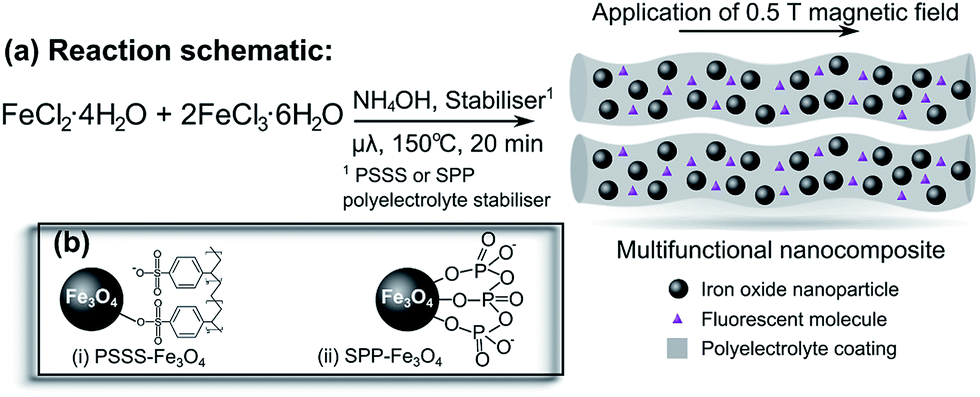 | ||
| Fig. 1 (a) Schematic representation of the one-pot synthesis of the multifunctional nanocomposite material, using microwave irradiation. Application of an external 0.5 T magnetic field may induce linear assemblies. Legend shows iron oxide nanoparticles, a fluorescent molecule (in this case, Rhodamine B), and a polyelectrolyte coating which make up this nanocomposite. (b) Binding of (i) PSSS and (ii) SPP stabilisers to the iron oxide nanoparticle surface. | ||
2 Results and discussion
A modified co-precipitation technique has been employed, wherein a precursor solution of ferric and ferrous chlorides in the presence of polyelectrolyte solution was first prepared. The co-precipitation method involves several processes: nucleation, seed formation and growth.50,51 Rapid particle nucleation follows the addition of ammonia base, after which the particle suspension is transferred to a microwave cavity and particle growth occurs under microwave irradiation at 150 °C for 20 min. All samples were washed until neutral. In the case of the bare particles, the sample was dried for further analysis. For the polyelectrolyte samples, the final, neutral fifth washings were highly stable colloidal suspensions, which have been characterised using dynamic light scattering (DLS), transmission electron microscopy (TEM), and MR imaging, and their cellular interactions were analysed. The remaining solids were dried and analysed using X-ray diffraction (XRD), X-ray absorption spectroscopy (XAS), SQUID magnetometry, thermogravimetric analysis (TGA) and IR.XRD patterns collected on dried powders of each sample are shown in Fig. 2(a). For comparison, a diffraction pattern was also collected of magnetite nanoparticles prepared using the traditional co-precipitation route without any microwave treatment (red line). The patterns obtained for all samples may be indexed to the cubic spinel, magnetite (Fe3O4). The broad peaks observed are typical for nanoparticles and the particle sizes, which may be obtained from the Scherrer equation, are included in Table 1. These are on the order of 10 nm. Also shown in Fig. 2 are the patterns obtained for polyelectrolyte-stabilised samples, labelled PSSS–Fe3O4 and SPP–Fe3O4. Given that the XRD patterns for magnetite and maghemite are similar, Raman spectroscopy measurements were performed. Using Raman spectroscopy, it is possible to assign characteristic peaks for magnetite (which contains both Fe3+ and Fe2+ ions) and the oxidised iron oxides maghemite (γ-Fe2O3) and haematite (α-Fe2O3). Fig. 2(b) shows the resulting Raman spectrum for the pure uncoated nanoparticles, displaying a strong peak at 664 cm−1 which can be attributed to the A1g mode of magnetite. Also visible are the Eg and T2g modes, centered at 301 and 527 cm−1 respectively. All peaks observed are in excellent agreement with previous assignments for magnetite in the literature.4,52,53 There are no peaks for haematite or maghemite observed.
 | ||
| Fig. 2 (a) Powder XRD patterns for samples prepared using microwave-assisted methods: bare magnetite (orange), PSSS–Fe3O4 (navy) and SPP–Fe3O4 (purple). For comparison, a pattern of magnetite nanoparticles prepared using traditional co-precipitation synthesis is included (red) together with a reference ICSD pattern. (b) Raman spectrum of pure Fe3O4 nanoparticles prepared using microwave-assisted method. Peak at 664 cm−1 is assigned to A1g mode of magnetite, with Eg and T2g modes centered at 301 and 527 cm−1 respectively. No peaks for haematite or maghemite were observed. | ||
| Sample | XRD (size nm) | TEM (size nm) | DLS Z-ave (PDI) | Zeta potential (mV) |
|---|---|---|---|---|
| a No measurements were carried out on pure Fe3O4 nanoparticles prepared using microwave methods as no stable suspension resulted. | ||||
| Fe3O4 | 11.2 | 12 ± 2 | N/A (N/A) | N/Aa |
| Fe3O4–PSSS | 16.1 | 13.4 ± 1.5 | 94.74 (0.097) | −41.5 |
| Fe3O4–SPP | 10.9 | 10.1 ± 1.5 | 104.93 (0.158) | −46.9 |
The comparatively broader peak shapes for the Fe3O4–SPP sample are reflected in the decreased particle size values obtained from the Scherrer equation for these particles (10.9 nm for SPP-coated particles, 11.2 nm for uncoated nanoparticles). HRTEM images for pure Fe3O4 samples reveal single-crystal nanoparticles (see Fig. 3). The Fe3O4 particles are aggregated, with a typical particle size of 12 ± 2 nm (measured for N = 100 particles). Analysis of lattice spacing and SAED patterns, shown in Fig. 3(d), confirm the nanoparticles to be magnetite (Fe3O4, JCPDS index card number 19-629). It is clear from these measurements that the microwave iron oxide sample contains both Fe2+ and Fe3+ ions, in good agreement with the Rietveld profile analysis of this sample to Fe3O4.
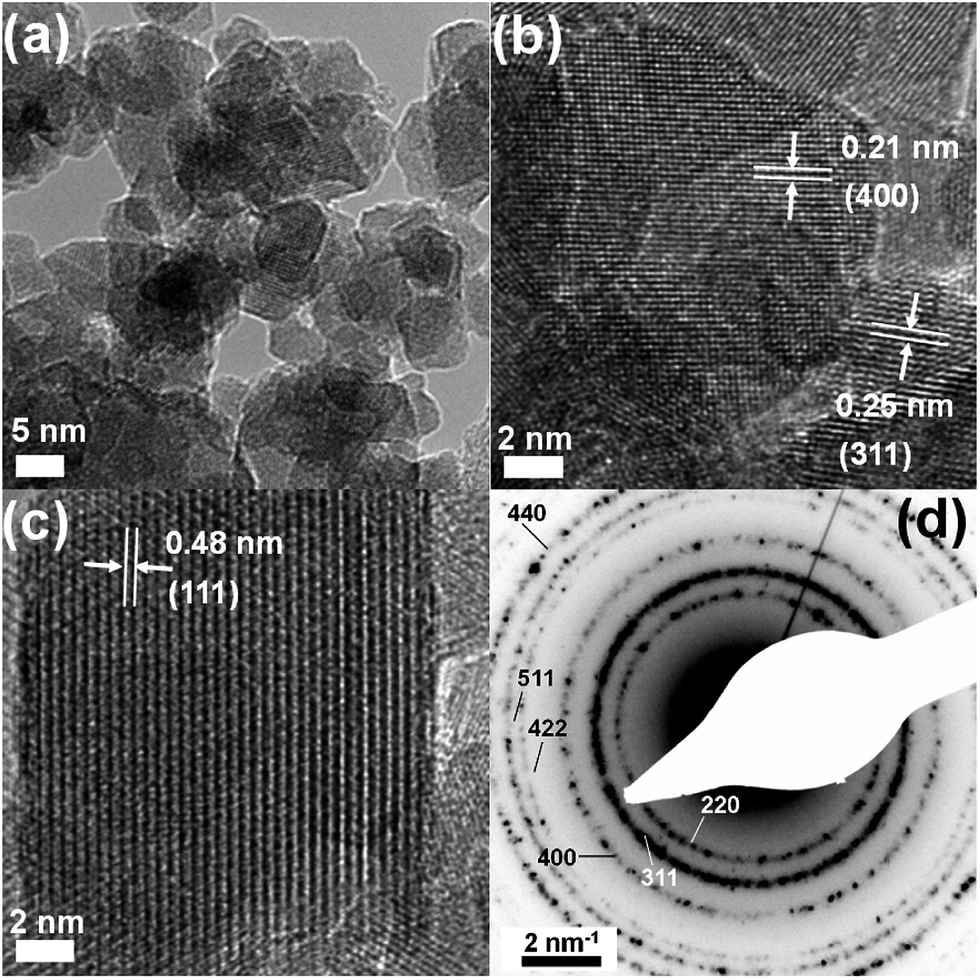 | ||
| Fig. 3 (a) HRTEM images of bare Fe3O4 nanoparticles prepared by microwave routes reveal aggregation of discrete nanoparticles. Each individual particle is highly crystalline, with lattice fringes clearly visible. (b and c) The interplanar spacings are indexed to the corresponding magnetite (hkl) reflection. (d) SAED pattern from an agglomerate of particles, with the pattern indexed to magnetite. | ||
Interestingly, the nanoparticle shape, size and aggregate nature are significantly affected by the polyelectrolyte stabilisers employed under otherwise identical reaction conditions. Electron microscopy images of the polyelectrolyte-stabilised samples are shown in Fig. 4, where the presence of the PSSS-polyelectrolyte is evidenced by the core–shell appearance of the nanocomposite (Fig. 4(a)). The presence of polyelectrolyte on the PSSS– and SPP–Fe3O4 nanoparticle surface has also been confirmed using IR spectroscopy (see ESI Fig. S1†). Both samples give an Fe–O stretch at ∼530 cm−1 and a broad stretch at 3400 cm−1 for physically adsorbed water on the particle surface. In the case of PSSS–Fe3O4, an Fe–O–S stretch is noted at 669 cm−1, which indicates that the mode of binding is through the sulfonate group to the surface iron atoms of the nanoparticles. There are also stretches noted for the sulfonate groups at 775, 830, 1115, 1160 and 1405 cm−1. In the case of the SPP–Fe3O4 sample, an Fe–O–P stretch is observed at 992 cm−1, indicating that the mode of binding is via the phosphate groups to the surface iron atoms. Stretches relating to the phosphate groups are also observed at 869 and 1255 cm−1. TGA plots for bare magnetite, PSSS– and SPP–Fe3O4 nanoparticles are shown in ESI Fig. S2a.† The mass loss for pure magnetite is low at 3.4% and likely represents the loss of strongly adsorbed water and dehydration of surface hydroxyl groups. The mass losses are greater for the PSSS– and SPP–Fe3O4 samples, at 9.3% and 7.8% respectively, due to the removal of the polyelectrolyte surfactant at increasing temperatures. For both polyelectrolytes, the particles appear aggregated in the electron microscopy images (Fig. 4), with higher magnification images clearly showing each agglomerated region consisting of numerous discrete nanoparticles, which are clustered together. The shape of pure Fe3O4 and PSSS–Fe3O4 nanoparticles appear better defined than the SPP–Fe3O4 sample, whose particles appear smaller on average and irregular in shape (Fig. 4(f)). A high degree of crystallinity of these polyelectrolyte-stabilised nanoparticles is observed, where lattice spacings consistent with magnetite are identified. SAED patterns are in excellent agreement with these observations (shown in ESI Fig. S3†).
 | ||
| Fig. 4 HRTEM images of (a and b) PSSS– and (d and e) SPP–Fe3O4 nanoparticles. The major difference here is the irregular particle shape of the SPP–Fe3O4 (f) compared to PSSS–Fe3O4 which has a core–shell appearance. The formation of regular, linear assemblies is noted for the PSSS–Fe3O4 samples (c) upon application of a 0.5 T magnetic field. | ||
In order to probe the behaviour of these polyelectrolyte nanocomposites in the presence of a magnetic field, samples were dried in a 0.5 T magnetic field and analysed using microscopy. In the case of the PSSS-stabilised nanoparticles, linear assemblies are observed (Fig. 4(c)); reminiscent of previous reports for samples prepared by co-precipitation alone.48 Currently, we are focussing attention on understanding how the nature of the polyelectrolyte and the chain length affect the formation of these linear assemblies and the resulting implications on imaging behaviour of nanoparticulate contrast agents.
The aggregation of these stabilised nanoparticles has been investigated further by measuring the average hydrodynamic radii at 298 K using DLS (ESI Fig. S2(b)†). For both samples and for a range of reproduced reactions, this average is found to be close to 100 nm. The polydispersity of the SPP–Fe3O4 sample is greater than that for the PSSS–Fe3O4 sample, which is evidenced by the broader peak shape and the slightly higher polydispersity index (PDI) (0.158 for SPP–Fe3O4 and 0.097 for PSSS–Fe3O4). The low values obtained for the PDI values (<0.2) indicate a unimodal distribution of monodisperse clusters for both samples. Upon exposure to a 0.5 T magnetic field these values do not change, confirming the excellent water stability these polyelectrolyte stabilisers confer on the Fe3O4 nanoparticles prepared using microwave methods. Table 1 summarises the particle sizes as calculated from Scherrer broadening (XRD), average particle size (TEM), hydrodynamic radius and zeta potential (DLS). The observed zeta potentials are below −30 mV for both samples, which confirms the negative surface charge we postulate in Fig. 1 and reaffirms the excellent water stability of these suspensions, which was the case over a six month period as confirmed by DLS analysis.54
Magnetization curves were measured at 300 K and 10 K in a magnetic field of up to 2 × 104 G and are shown in Fig. 5. There is negligible coercivity and remanence noted at 300 K, indicative of superparamagnetic, single-domain iron oxide particles. The magnetization is unsaturated up to 2 T, even at 10 K. Uncoated magnetite prepared using our microwave-assisted method gives a saturation magnetisation of 67.6 emu g−1 at 300 K. While this is lower than the theoretical value for bulk magnetite of 98 emu g−1 (most likely due to spin disorder on the particle surface55), this value is higher than previously reported saturation magnetisation values for aqueous routes to iron oxide nanoparticles (40–50 emu g−1).56,57 By employing polyelectrolytes as stabilisers we have found that primary particle sizes and morphologies change and this, in turn, has a marked effect on the resulting magnetic properties. The SPP-stabilised particles are smaller in size, with ill-defined shape, and this is reflected in the reduced Ms value of 53.9 emu g−1 at 300 K. The PSSS-stabilised samples, on the other hand, have an Ms value of 77.1 emu g−1 at 300 K and appear highly crystalline in HRTEM. Interestingly, the magnetisation value here is significantly higher than Ms values previously obtained from NMRD data for PSSS-stabilised Fe3O4 (30–50 emu g−1) prepared without the additional microwave synthesis step.58
 | ||
| Fig. 5 Magnetic hysteresis loops of pure Fe3O4, PSSS–Fe3O4 and SPP–Fe3O4 at (a) 300 K and (b) 10 K. Data are shown per gram of Fe3O4 which was determined from TGA results. | ||
The saturation magnetisation value is greatly affected by the crystallinity of the sample. For high temperature decomposition routes using organic surfactants, for example, the highly crystalline and uniform nature of the particles is manifested in similarly higher Ms values (∼80 emu g−1).59 For biological use, organic-surfactant coated particles require further post-processing to transfer into aqueous solutions to obtain stable suspensions.26,60 The microwave-assisted synthesis reported here achieves both properties in a single step: excellent magnetic properties combined with long-term aqueous stability. From these data, we have shown the combination of the PSSS stabiliser and microwave irradiation results in a water-stable crystalline material, without compromising the magnetic properties.
To evaluate the MR efficacy of the PSSS–Fe3O4 and SPP–Fe3O4 samples, T1, T2 and T*2-weighted images were collected at varying Fe concentrations using a 3 T clinical MRI scanner. Relaxivity values are reported in Table 2. Increasing Fe concentrations (∼0–2 mM in H2O) were imaged and are displayed in Fig. 6. Higher relaxation rates are noted upon increasing Fe concentration, because PSSS–Fe3O4 and SPP–Fe3O4 shorten the T2 relaxation time, which reduces the signal intensity, i.e. negative contrast. Interestingly, the contrast displayed by the PSSS–Fe3O4 sample is greater than that of the SPP–Fe3O4 particles. The increased crystallinity of these particles and their enhanced magnetic properties are the major factors for this behaviour. Since it is the magnetic moments of the particles interacting with the water protons that result in image contrast, the crystalline nature of the particles is vital in determining how efficiently this may occur. In the case of the SPP-stabilised nanoparticles, the irregular shape and decrease in saturation magnetization point to greater disorder of the surface spins in these particles which will play a role in decreasing the imaging efficacy. This is in good agreement with an extensive study reported by Vuong et al., who have proposed a method for predicting T2 relaxation based on the nanocrystal size and magnetization values.61 For our PSSS–Fe3O4 particles, the excellent magnetic properties and the crystalline nature of the primary particle indicate the promising potential for these contrast agents for MR imaging. MR contrast properties for several other reports of functionalised iron oxide nanoparticles are also included in Table 2.48,62,63
| Sample | Field | r1 (mM−1 s−1) | r2 (mM−1 s−1) | r*2 (mM−1 s−1) | r2/r1 |
|---|---|---|---|---|---|
| a Note these measurements performed in 1.5 T field. | |||||
| Fe3O4–PSSS | 3 T | 3.18 ± 0.10 | 26.02 ± 6.54 | 179.00 ± 22.75 | 8.18 |
| Fe3O4–SPP | 3 T | 2.74 ± 0.51 | 17.04 ± 0.91 | 179.31 ± 10.66 | 6.21 |
| Fe3O4–PSSS (ref. 48)a | 1.5 T | 7.2 | 89.4 | — | 0.08 |
| Fe3O4–PEG–BP (ref. 62) | 3 T | 9.5 | 28.2 | — | 2.97 |
| Fe3O4–PEG (ref. 63) | 3 T | 4.77 | 29.2 | — | 6.12 |
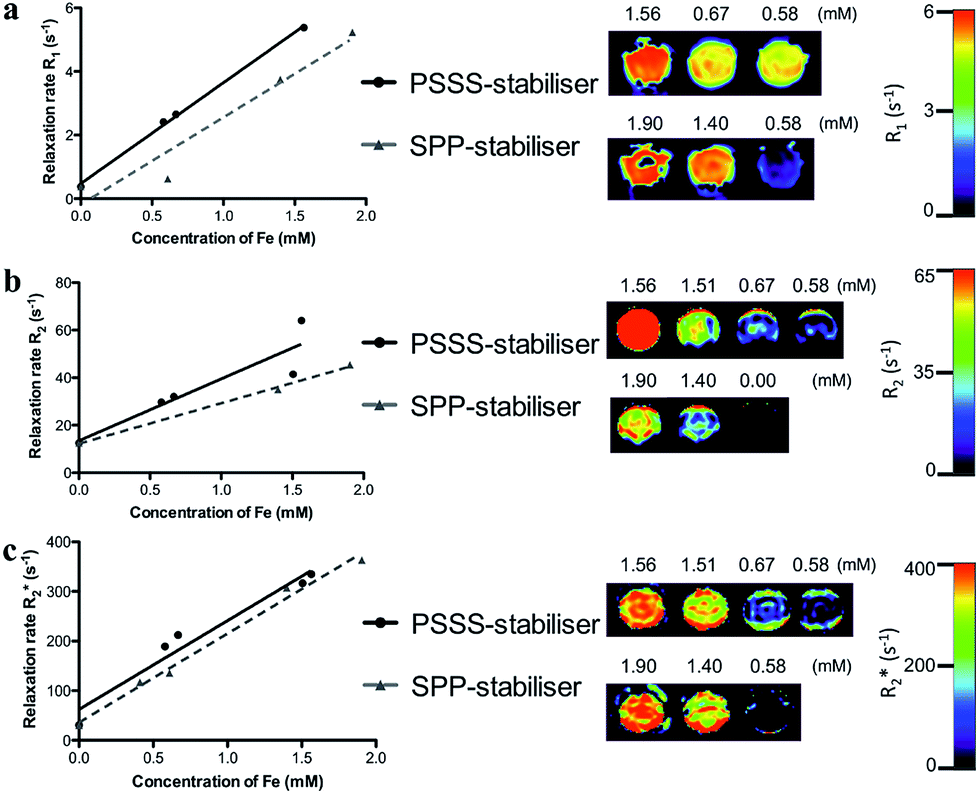 | ||
| Fig. 6 Relaxivities of the PSSS (straight line) and SPP (dashed line) nanocomposites measured at 3 T and 20 °C. Scatter plots show correlations between measured R1 (a), R2 (b), and R*2 (c) values of the nanocomposites and iron concentrations measured using ICP-MS. The relaxation rates (R1, R2 and R*2) were determined at 3 T using T1, T2 and T*2 mapping sequences, respectively, and aqueous solutions between 0 and 2 mM of the contrast agents. Pearson’s correlation coefficient values ranged from 0.92 to 0.99. Phantom MRI images of the formulations show R1, R2 and R*2 maps in colour scale. R1, R2 and R*2 values increase with increasing concentrations of contrast agents (highest concentration on the left). | ||
Enhanced MR contrast has been previously noted for linear assemblies of iron oxide nanoparticles.48,58,64 In the current case, we observe relaxivities on the order of commercially available contrast agents and nanoparticle suspensions prepared by high temperature decomposition routes, with the added advantage here of no additional work-up required to transfer to aqueous conditions.65–67 Recently, Zboril and co-workers reported extraordinarily high relaxivities of 735 mM s−1 for iron oxide nanoparticles with a 1 nm terephthalic acid coating, postulating that effective spin-transfer to surrounding water protons is mediated via π-conjugation pathways through the organic surfactant.68 It is enticing to consider the combined use of active spin-transfer surfactants with microwave processing to further optimise relaxivity behaviour.
The effect of stabiliser on cell toxicity was examined using cell viability studies on suspensions of the polyelectrolyte-stabilised particles co-incubated at increasing concentrations (0.001–1000 μg L−1) with a range of cell lines and these results are depicted in Fig. 7. Regardless of the stabiliser employed, all nanoparticle suspensions tested were found to be non-toxic to a variety of mammalian cell lines, demonstrating the non-toxic robustness of these materials. Iron concentration ranges were similar to previous reports.69 This is determined by the fact they did not affect the viability of UKF-NB-3 neuroblastoma cells, primary human retinal pigment epithelial (RPE) cells, or primary human foreskin fibroblasts (HFF) even in concentrations up to 1 mg of iron per L.
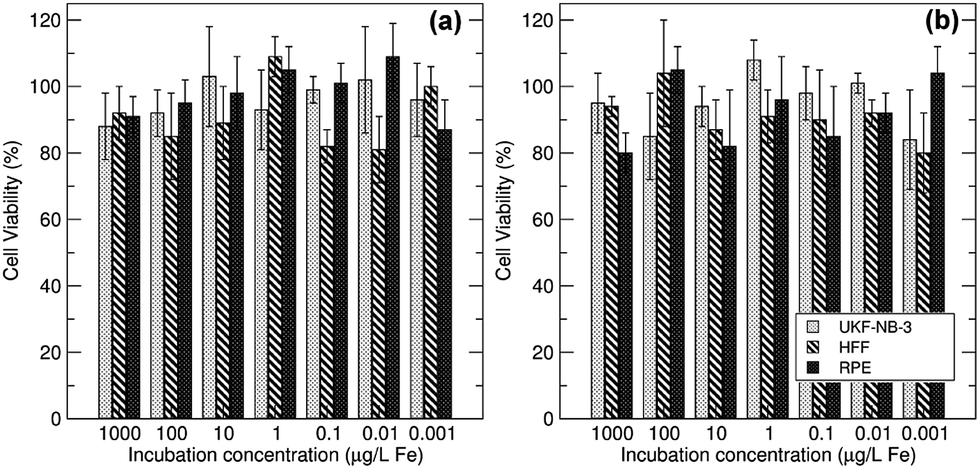 | ||
| Fig. 7 Cytotoxicity profiles of (a) PSSS–Fe3O4 and (b) SPP–Fe3O4 in three different cell lines of UKF-NB-3 neuroblastoma cells, primary human retinal pigment epithelial (RPE) cells and primary human foreskin fibroblasts (HFF) in concentrations up to 1 mg iron per L. | ||
To demonstrate uptake into mammalian cells, fluorescently-labelled samples were prepared by the addition of Rhodamine B to the PSSS polyelectrolyte solution before particle precipitation, where association is driven by the electrostatic interactions between the dye and the polyelectrolyte. The fluorescent nature of the nanocomposite was confirmed using fluorescence spectroscopy (λex = 545 nm) and an enhanced aggregation of the composite is noted in DLS, where electrostatic interactions drive the formation of larger cluster sizes and this is reflected by the larger Z-ave (108.2 nm; PDI 0.169) (see ESI Fig. S4†). The sample spent several days under a 0.5 T magnetic field before DLS measurement to remove any larger aggregates and to ensure a stable suspension remained. Removal of any excess dye was confirmed by continuous washing of the sample and evaluation using fluorescence spectroscopy before redispersion into water and also by dialysis experiments of the resulting nanocomposite (see ESI Fig. S5†). Fig. 8 shows the resulting nanoparticle uptake by UKF-NB-3 neuroblastoma cells, which can be clearly located throughout the cytoplasm. Of importance here is that this internalisation process had no effect on viability as described above. Complementary Z-scan images confirm the location of the functionalised nanoparticles throughout the cell cytoplasm (Fig. 8(c)), rather than on the cell surface.
 | ||
| Fig. 8 Immunofluorescence confocal microscopy imaging of UKF-NB-3 neuroblastoma cells treated with nanoparticles for 24 h. In all cells, the nanoparticle suspension was internalised (a) and particles appear punctated (b). Internalisation is confirmed when viewing slices through the cell (c). | ||
3 Conclusions
In conclusion, we have demonstrated a reproducible and reliable single step route to multifunctional magnetic nanocomposite materials, using a microwave-assisted synthesis, in order to provide stable magnetic fluids for use as MR contrast agents. Here, the underpinning structure–property relationship in these materials is highlighted by the synthetic approach taken and the resulting outcome this has on the functional properties of the nanocomposites. The choice of stabilizer – polysulfate versus polyphosphate – has a substantial effect on the magnetic properties, which translates to their ability to enhance the relaxation mechanism of surrounding water protons in MR imaging. Whilst not the highest relaxivities reported for iron oxide nanocomposites, the comprehensive analysis presented here using a full range of characterisation techniques demonstrates PSSS as an excellent stabiliser for the preparation of multifunctional magnetic nanocomposite materials and our microwave-assisted synthetic approach can be applied to a full range of ferrite-based nanoparticles and polyelectrolyte-stabilised systems. Previous reports have shown that effective contrast agents can be obtained using PSSS as a stabiliser for iron oxide nanoparticles without any microwave treatment.48,58 In the case of the nanocomposites prepared without microwave heating, the hydrodynamic radius and polydispersity index is greater (136 nm and 0.21, respectively). Most interesting though is that the saturation magnetisation (obtained from NMR dispersion measurements) is consistently lower for samples prepared without the application of microwave heating (30–50 emu g−1). This has a resulting implication on the contrast agent efficacy (values of r1 and r2 obtained are of the same order for both samples, but for the sample prepared without microwave treatment, the relaxation properties were measured only at 1.5 T). Therefore, the addition of this microwave heating step in the case of the PSSS stabiliser positively affects the subsequent properties and functionality of the nanocomposite. The direct link between particle crystallinity and the resulting magnetic behaviour and the governing effect this has on MR imaging capability is particularly interesting. We show that these nanocomposites are non-toxic to a range of mammalian cells, where their uptake can be confocally imaged. The ease of this approach allows for the preparation of extremely stable magnetic fluids for combined MR contrast efficacy and optical imaging and paves the way for a synthetic methodology which allows for greater control over final functionality.4 Experimental
4.1 Materials
All materials were obtained from Sigma Aldrich. Millipore water was deoxygenated by boiling and then cooling under nitrogen gas. A CEM Discover SP system was used for microwave heating. FTIR spectra (400–2000 cm−1) were recorded using a Shimadzu IR Affinity-1 spectrophotometer. X-ray diffraction was performed on a Bruker D8 X-ray diffractometer and powder patterns were analysed using Rietveld refinement as embodied in the Fullprof suite. Thermogravimetric analysis was obtained with a Netzsch STA 409 PC Luxx TGA machine. All samples were heated in air to 700 °C. Dynamic light scattering measurements were performed using a Malvern Zetasizer nano-ZS. Transmission electron microscopy (TEM) images were taken on a JEOL JEM (200-FX) operating at 120 kV. Samples were prepared on a formvar coated copper grid. Some grids were dried over a 0.5 T magnet. High resolution TEM and selected area electron diffraction (SAED) were performed on an FEI Tecnai TF20 instrument fitted with a field emission gun, operated at 200 keV. TEM samples were prepared by dispersing the sample in deionised water and dropping the solution onto an amorphous holey carbon coated grid. TEM data were obtained and processed using either Digital Micrograph or IMAGEJ 1.41 software.4.2 Preparation of magnetic nanocomposites
FeCl3·6H2O (2.70 g; 10 mmol) and FeCl2·4H2O (0.99 g; 5 mmol) were dissolved in 10 mL of deoxygenated water. Polyelectrolyte stabiliser (0.2 g of either PSSS or SPP) was dissolved in the iron solution. The solution was heated to 80 °C. Ammonia solution (10 mL; 28–30%) was injected at a rate of approximately 2.5 mL s−1 and the solution was stirred for 20 minutes before transferring to the microwave cavity to be heated at 150 °C for 20 minutes. For fluorescently-labelled samples, Rhodamine B (2 × 10−4 g; 10 mmol) was dissolved in 2 mL of deoxygenated water and the PSSS polyelectrolyte (0.2 g) was added to this and stirred for two hours. This was transferred to 10 mL of Fe3+/Fe2+ solution (2.70 g; 10 mmol, and 0.99 g; 5 mmol respectively), before addition of 10 mL of ammonia solution at 80 °C. The resulting black precipitates were washed with Millipore water (5 × 20 mL) with the final two washings being used in TEM, DLS, FTIR, MRI and confocal measurements. The solid precipitate was analysed with XRD, TGA, HRTEM and SQUID measurements.4.3 MR imaging
The tubes of different PSSS–Fe3O4 and SPP–Fe3O4 concentrations were placed in a rack in the centre of the magnet. MR imaging was performed with a standard extremity flex coil on a clinical 3 T MRI scanner (Achieva, Philips Healthcare, Best, The Netherlands). T2 was determined with a 2D multi-spin-echo sequence (FOV = 120 × 120 mm2, matrix = 432 × 432, measured slice thickness = 3 mm, echo train length = 5, TE = 10 ms, TR = 725 ms, flip angle = 90°). The acquired imaging data was transferred to a computer running Matlab and analysed using an in-house Matlab tool to receive the relaxation time T2 for each Fe concentration. Excel was used to plot the relaxation rate R2 over the concentration and the relaxivity value was determined using linear regression. Iron concentrations of all MRI scanned serial dilutions of SPIONs were determined using Inductively Coupled Plasma Mass Spectrometry (ICP-MS). Briefly, samples were digested in 70% nitric acid overnight at room temperature, followed by dilution in deionized water. A standard curve was acquired with each sample set for iron concentration determination.4.4 Investigating the effect of nanoparticles on cell viability
The effects of the nanoparticles on cell viability were determined in UKF-NB-3 neuroblastoma cells, primary human retinal pigment epithelial cells, and primary human foreskin fibroblast cells. UKF-NB-3 cells were cultivated in IMDM supplemented with 10% foetal calf serum (FCS), 100 IU mL−1 penicillin, and 100 mg mL−1 streptomycin. Retinal pigment epithelial cells were cultured in DMEM supplemented with 20% FCS, 100 IU mL−1 penicillin, and 100 mg mL−1 streptomycin. Fibroblasts were cultured in DMEM supplemented with 10% FCS, 100 IU mL−1 penicillin, and 100 mg mL−1 streptomycin. All cells were cultivated at 37 °C in humidified 5% CO2 atmosphere as previously described.70,71 Cell viability upon the addition of nanoparticle preparations at different concentrations was determined by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) dye reduction assay after 120 h of incubation as described previously.70,714.5 Determination of cellular location of nanoparticles using fluorescence microscopy
UKF-NB3 cells were seeded at 2 × 105 cells per well in a 24 well plate, containing a coverslip in each well, and grown at 37 °C for 48 hours. Rhodamine conjugated nanoparticles were diluted to 0.099 mg L−1 iron concentration in media, 1 mL added to appropriate wells and incubated for 3 hours before aspirating and fixing with methanol at −20 °C for 5 minutes. Coverslips were mounted using mowiol and anti-fade and slides examined using a Leica confocal laser scanning (TCS 4; ×63 oil lens) microscope.Acknowledgements
The authors gratefully acknowledge Prof. Ram Seshadri at UCSB for help, encouragement and for the use of SQUID facilities. These measurements were made possible through funding from the Royal Society of Chemistry Journal Grant for International Authors provided to SAC. SAC acknowledges funding by the EPSRC (EP/K029290/1) and the Royal Society (RG100301). SAC, ES and ERA gratefully acknowledge funding from the Leverhulme Trust (RPG-2015-134). RTMR is funded by The Centre of Excellence in Medical Engineering funded by the Wellcome Trust and EPSRC under Grant No. WT 088641/Z/09/Z. The authors acknowledge financial support from the Department of Health via the National Institute for Health Research (NIHR) comprehensive Biomedical Research Centre award to Guy’s & St Thomas’ NHS Foundation Trust in partnership with King’s College London and King’s College Hospital NHS Foundation Trust.References
- Q. Zeng, I. Baker, J. A. Loudis, Y. Liao, P. Hoopes and J. B. Weaver, Appl. Phys. Lett., 2007, 90, 233112 CrossRef.
- Q. A. Pankhurst, J. Connolly, S. K. Jones and J. Dobson, J. Phys. D: Appl. Phys., 2009, 42, 224001 CrossRef.
- C. Sun, J. S. Lee and M. Zhang, Adv. Drug Delivery Rev., 2008, 60, 1252–1265 CrossRef CAS PubMed.
- L. Ternent, D. A. Mayoh, M. R. Lees and G.-L. Davies, J. Mater. Chem. B, 2016, 4, 3065–3074 RSC.
- H. B. Na, I. C. Song and T. Hyeon, Adv. Mater., 2009, 21, 2133–2148 CrossRef CAS.
- M. J. Williams and S. A. Corr, Frontiers of Nanoscience, Elsevier, 2013, pp. 29–63 Search PubMed.
- A.-H. Lu, E. L. Salabas and F. Schüth, Angew. Chem., Int. Ed., 2007, 46, 1222–1244 CrossRef CAS PubMed.
- Y.-X. J. Wang, Quant. Imag. Med. Surg., 2011, 1, 35–40 Search PubMed.
- D. Yoo, J.-H. Lee, T.-H. Shin and J. Cheon, Acc. Chem. Res., 2011, 44, 863–874 CrossRef CAS PubMed.
- L. H. Reddy, J. L. Arias, J. Nicolas and P. Couvreur, Chem. Rev., 2012, 112, 5818–5878 CrossRef CAS PubMed.
- J.-H. Park, G. von Maltzahn, M. J. Xu, V. Fogal, V. R. Kotamraju, E. Ruoslahti, S. N. Bhatia and M. J. Sailor, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 981–986 CrossRef CAS PubMed.
- Q. Song and Z. J. Zhang, J. Phys. Chem. B, 2006, 110, 11205–11209 CrossRef CAS PubMed.
- R. Ladj, A. Bitar, M. Eissa, Y. Mugnier, R. Le Dantec, H. Fessi and A. Elaissari, J. Mater. Chem. B, 2013, 1, 1381–1396 RSC.
- C. N. R. Rao, H. S. S. R. Matte, R. Voggu and A. Govindaraj, Dalton Trans., 2012, 41, 5089–5120 RSC.
- G. R. Patzke, Y. Zhou, R. Kontic and F. Conrad, Angew. Chem., Int. Ed., 2011, 50, 826–859 CrossRef CAS PubMed.
- R. Massart, IEEE Trans. Magn., 1981, 1247–1248 CrossRef CAS.
- D. Kim, Y. Zhang, W. Voit, K. Rao and M. Muhammed, J. Magn. Magn. Mater., 2001, 225, 30–36 CrossRef CAS.
- S. Ge, X. Shi, K. Sun, C. Li, C. Uher, J. R. Baker Jr, M. M. B. Holl and B. G. Orr, J. Phys. Chem. C, 2009, 113, 13593–13599 CAS.
- S. Sun and H. Zeng, J. Am. Chem. Soc., 2002, 124, 8204–8205 CrossRef CAS PubMed.
- Y. Tian, B. Yu, X. Li and K. Li, J. Mater. Chem., 2011, 21, 2476–2481 RSC.
- A. G. Roca, M. P. Morales, K. O’Grady and C. J. Serna, Nanotechnology, 2006, 17, 2783 CrossRef CAS.
- S. Laurent, D. Forge, M. Port, A. Roch, C. Robic, L. V. Elst and R. N. Muller, Chem. Rev., 2008, 108, 2064–2110 CrossRef CAS PubMed.
- I. Bilecka and M. Niederberger, Nanoscale, 2010, 2, 1358–1374 RSC.
- J. A. Gerbec, D. Magana, A. Washington and G. F. Strouse, J. Am. Chem. Soc., 2005, 127, 15791–15800 CrossRef CAS PubMed.
- T. E. Ashton, J. V. Laveda, D. A. MacLaren, P. J. Baker, A. Porch, M. O. Jones and S. A. Corr, J. Mater. Chem. A, 2014, 2, 6238–6245 CAS.
- O. Pascu, E. Carenza, M. Gich, S. Estradé, F. Peiró, G. Herranz and A. Roig, J. Phys. Chem. C, 2012, 116, 15108–15116 CAS.
- C. Blanco-Andujar, D. Ortega, P. Southern, Q. A. Pankhurst and N. T. K. Thanh, Nanoscale, 2015, 7, 1768–1775 RSC.
- R. M. Wong, D. A. Gilbert, K. Liu and A. Y. Louie, ACS Nano, 2012, 6, 3461–3467 CrossRef CAS PubMed.
- E. A. Osborne, T. M. Atkins, D. A. Gilbert, S. M. Kauzlarich, K. Liu and A. Y. Louie, Nanotechnology, 2012, 23, 215602 CrossRef PubMed.
- I. Bilecka, I. Djerdj and M. Niederberger, Chem. Commun., 2008, 7, 886–888 RSC.
- S. Komarneni, R. Roy and Q. H. Li, Mater. Res. Bull., 1992, 27, 1393–1405 CrossRef CAS.
- Q. Li and Y. Wei, Mater. Res. Bull., 1998, 33, 779–782 CrossRef CAS.
- X. Hu, J. C. Yu, J. Gong, Q. Li and G. Li, Adv. Mater., 2007, 19, 2324–2329 CrossRef CAS.
- X. Hu and J. C. Yu, Adv. Funct. Mater., 2008, 18, 880–887 CrossRef CAS.
- V. Sreeja and P. Joy, Mater. Res. Bull., 2007, 42, 1570–1576 CrossRef CAS.
- R. Y. Hong, T. T. Pan and H. Z. Li, J. Magn. Magn. Mater., 2006, 303, 60–68 CrossRef CAS.
- E. Carenza, V. Barcelò, A. Morancho, J. Montaner, A. Rosell and A. Roig, Acta Biomater., 2014, 10, 3775–3785 CrossRef CAS PubMed.
- J. Huang, H. Pen, Z. Xu and C. Yi, React. Funct. Polym., 2008, 68, 332–339 CrossRef CAS.
- F. Bensebaa, F. Zavaliche, P. L’Ecuyer, R. W. Cochrane and T. Veres, J. Colloid Interface Sci., 2004, 277, 104–110 CrossRef CAS PubMed.
- S. Komarneni, M. C. D’Arrigo, C. Leonelli, G. C. Pellacani and H. Katsuki, J. Am. Ceram. Soc., 1998, 81, 3041–3043 CrossRef CAS.
- S. A. Corr, Y. P. Rakovich and Y. K. Gun’ko, Nanoscale Res. Lett., 2008, 3, 87–104 CrossRef CAS.
- A. Guerrero-Martínez, J. Pérez-Juste and L. M. Liz-Marzán, Adv. Mater., 2010, 22, 1182–1195 CrossRef PubMed.
- V. Salgueirino-Maceira and M. A. Correa-Duarte, Adv. Mater., 2007, 19, 4131–4144 CrossRef CAS.
- F. M. Kievit and M. Zhang, Acc. Chem. Res., 2011, 44, 853–862 CrossRef CAS PubMed.
- M. Liong, J. Lu, M. Kovochich, T. Xia, S. G. Ruehm, A. E. Nel, F. Tamanoi and J. I. Zink, ACS Nano, 2008, 2, 889–896 CrossRef CAS PubMed.
- S. Liu, F. Lu, X. Jia, F. Cheng, L.-P. Jiang and J.-J. Zhu, CrystEngComm, 2011, 13, 2425–2429 RSC.
- S. J. Byrne, S. A. Corr, Y. K. Gun’ko, J. M. Kelly, D. F. Brougham and S. Ghosh, Chem. Commun., 2004, 22, 2560–2561 RSC.
- S. A. Corr, S. J. Byrne, R. Tekoriute, C. J. Meledandri, D. F. Brougham, M. Lynch, C. Kerskens, L. O’Dwyer and Y. K. Gun’ko, J. Am. Chem. Soc., 2008, 130, 4214–4215 CrossRef CAS PubMed.
- G.-L. Davies, S. A. Corr, C. J. Meledandri, L. Briode, D. F. Brougham and Y. K. Gun'ko, ChemPhysChem, 2011, 12, 772–776 CrossRef CAS PubMed.
- S. A. Corr, Nanoscience, 2013, 180–207 CAS.
- R. Viswanatha and D. D. Sarma, Nanomater. Chem., 2007, 139–170 CAS.
- O. N. Shebanova and P. Lazor, J. Raman Spectrosc., 2003, 34, 845–852 CrossRef CAS.
- L. Slavov, M. V. Abrashev, T. Merodiiska, C. Gelev, R. E. Vandenberghe, I. Markova-Deneva and I. Nedov, J. Magn. Magn. Mater., 2010, 322, 1904–1911 CrossRef CAS.
- D. Cunningham, R. E. Littleford, W. E. Smith, P. J. Lundahl, I. Khan, D. W. McComb, D. Graham and N. Laforest, Faraday Discuss., 2006, 132, 135–145 RSC.
- P. Guardia, A. Labarta and X. Batlle, J. Phys. Chem. C, 2011, 115, 390–396 CAS.
- M. Mikhaylova, D. K. Kim, N. Bobrysheva, M. Osmolowsky, V. Semenov, T. Tsakalakos and M. Muhammed, Langmuir, 2004, 20, 2472–2477 CrossRef CAS PubMed.
- A.-L. Morel, S. Nikitenko, K. Gionnet, A. Wattiaux, J. Lai-Kee-Him, C. Labrugere, B. Chevalier, G. Deleris, C. Petibois, A. Brisson and M. Simonoff, ACS Nano, 2008, 2, 847–856 CrossRef CAS PubMed.
- S. A. Corr, Y. K. Gun’ko, R. Tekoriute, C. J. Meledandri and D. F. Brougham, J. Phys. Chem. C, 2008, 112, 13324–13327 CAS.
- S. Sun, H. Zeng, D. B. Robinson, S. Raoux, P. M. Rice, S. X. Wang and G. Li, J. Am. Chem. Soc., 2004, 126, 273–279 CrossRef CAS PubMed.
- Y. Wang, J. F. Wong, X. Teng, X. Z. Lin and H. Yang, Nano Lett., 2003, 3, 1555–1559 CrossRef CAS.
- Q. L. Vuong, J.-F. Berret, J. Fresnais, Y. Gossuin and O. Sandre, Adv. Healthcare Mater., 2012, 1, 502–512 CrossRef CAS PubMed.
- L. Sandiford, A. Phinikaridou, A. Protti, L. K. Meszaros, X. Cui, Y. Yan, G. Frodsham, P. A. Williamson, N. Gaddum, R. M. Botnar, P. J. Blower, M. A. Green and R. T. M. de Rosales, ACS Nano, 2013, 7, 500–512 CrossRef CAS PubMed.
- B. H. Kim, N. Lee, H. Kim, K. An, Y. I. Park, Y. Choi, K. Shin, Y. Lee, S. G. Kwon, H. B. Na, J.-G. Park, T.-Y. Ahn, Y.-W. Kim, W. K. Moon, S. H. Choi and T. Hyeon, J. Am. Chem. Soc., 2011, 133, 12624–12631 CrossRef CAS PubMed.
- J.-H. Park, G. von Maltzahn, L. Zhang, M. P. Schwartz, R. Ruoslahti, S. N. Bhatia and M. J. Sailor, Adv. Mater., 2008, 20, 1630–1635 CrossRef CAS PubMed.
- J. Qin, S. Laurent, Y. Jo, A. Roch, M. Mikhaylova, Z. Bhujwalla, R. Muller and M. Muhammed, Adv. Mater., 2007, 19, 1874–1878 CrossRef CAS.
- Z. Li, P. W. Yi, Q. Sun, H. Lei, H. Li Zhao, Z. H. Zhu, S. C. Smith, M. B. Lan and G. Q. M. Lu, Adv. Funct. Mater., 2012, 22, 2387–2393 CrossRef CAS.
- C. Sun, K. Du, C. Fang, N. Bhattarai, O. Veiseh, F. Kievit, Z. Stephen, D. Lee, R. G. Ellenbogen, B. Ratner and M. Zhang, ACS Nano, 2010, 4, 2402–2410 CrossRef CAS PubMed.
- D. Maity, G. Zoppellaro, V. Sedenkova, J. Tucek, K. Safarova, K. Polakova, K. Tomankova, C. Diwoky, R. Stollberger, L. Machala and R. Zboril, Chem. Commun., 2012, 48, 11398–11400 RSC.
- U. S. Patil, S. Adireddy, A. Jaiswal, S. Mandava, B. R. Lee and D. B. Chrisey, Int. J. Mol. Sci., 2015, 26, 24417–24450 CrossRef PubMed.
- M. Michaelis, C. Paulus, N. Löschmann, S. Dauth, E. Stange, H. Doerr, M. Nevels and J. Cinatl, Cell. Mol. Life Sci., 2011, 68, 1079–1090 CrossRef CAS PubMed.
- M. Michaelis, F. Rothweiler, B. Agha, S. Barth, Y. Voges, N. Löschmann, A. von Deimling, R. Breitling, H. W. Doerr, F. Rödel, D. Speidel and J. Cinatl Jr, Cell Death Dis., 2012, 3, e294 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra11819d |
| This journal is © The Royal Society of Chemistry 2016 |