
Structure and electrical properties of lead-free Bi0.5Na0.5TiO3-based ceramics for energy-storage applications
Abstract
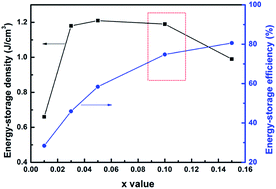
A new energy-storage ceramic system based on (1 − x)(Bi0.5Na0.5TiO3–BaTiO3)–xNaTaO3 ((1 − x)(BNT–BT)–xNT) is reported in this study. XRD refinement indicated a composition induced rhombohedral to tetragonal phase transition. All the samples exhibited a dense microstructure with an average grain size of 1.2–1.9 μm. The introduction of NT greatly improved the temperature stability of the dielectric properties for the BNT–BT system. For compositions x = 0.03–0.15, the working temperature range spanned over 260 °C satisfying TCC150 °C ≤ ±15%. The electric conductivity as a function of frequency followed the double power law. In the temperature region of 325–500 °C, the activation energy of DC conduction ranged from 1.47 eV to 1.71 eV, indicating intrinsic band-type electronic conduction. The optimum energy-storage properties were obtained in 0.90(BNT–BT)–0.10NT with an energy-storage density of 1.2 J cm−3 and energy-storage efficiency of 74.8% at 10 kV mm−1. The results demonstrate that (1 − x)(BNT–BT)–xNT ceramics are promising candidates for high-temperature energy-storage applications.
 Please wait while we load your content...
Please wait while we load your content...

