
Modified structure of two-dimensional polythiophene derivatives by incorporating electron-deficient units into terthiophene-vinylene conjugated side chains and the polymer backbone: synthesis, optoelectronic and self-assembly properties, and photovoltaic application†
Chuen-Yo Hsiow*ab,
Yu-Hsiang Linc,
Rathinam Rajaa,
Syang-Peng Rweic,
Wen-Yen Chiud,
Chi-An Daid and
Leeyih Wang*a
aCenter for Condensed Matter Sciences, National Taiwan University, 10617 Taipei, Taiwan. E-mail: leewang@ntu.edu.tw; chuenyo@caltech.edu
bJoint Center for Artificial Photosynthesis, California Institute of Technology, 91125 Pasadena, USA
cInstitute of Organic and Polymeric Materials, National Taipei University of Technology, 10608 Taipei, Taiwan
dDepartment of Chemical Engineering, National Taiwan University, 10617 Taipei, Taiwan
First published on 8th July 2016
Abstract
Molecular engineering on the conjugated side chains of two-dimensional (2D) conjugated polymers was conducted and its effect on the optical, electronic, self-assembly and photovoltaic properties was investigated. A new monomer, M2, was prepared by capping (E)-3′-(2-(2,5-dibromothiophen-3-yl)vinyl)-4,4′′-bis(2-ethylhexyl)-2,2′:5′,2′′-terthiophene, M1, with two heptanoyl groups, and then coupled with 5,5′-bis(trimethylstannyl)-2,2′-bithiophene via microwave-assisted Stille polymerization to produce a series of polythiophene derivatives with terthiophene-vinylene conjugated side chains, TTV–PTs. Copolymer P2 shows a down-shifted HOMO energy level, enhanced solubility, and red-shifted absorption, as compared with P1; however, the bulky side chains significantly disrupt the coplanarity of thiophene rings in the polymer backbone and the ability to self-assemble into an ordered structure. The GIXRD measurements reveal that the original crystallinity of P1 can be recovered by simply inserting a few 2,1,3-benzothiadiazole units into the polythiophene main chain in P2 through a random copolymerization route to yield a terpolymer, P3, which possesses excellent crystallinity, thereby causing a three-fold increment in hole mobility. Furthermore, the P1/PC61BM, P2/PC61BM, and P3/PC61BM solar devices exhibit power conversion efficiencies of 3.89%, 1.52%, and 4.17%, respectively, under AM1.5G illumination with an intensity of 100 mW cm−2.
Introduction
Molecular engineering of specific semiconducting conjugated polymers for use in high-performance organic electronic devices, such as field effect transistors (FETs), photo-detectors, light-emitting diodes (LEDs), and photovoltaics (PVs), requires a better understanding of the relationship between their chemical structure and optoelectronic properties.1–7 Several successful strategies for the structural design of conjugated polymers, such as donor–acceptor (D–A) copolymerization,8–10 two-dimensional (2-D) conjugation,11–13 and side-chain engineering,14–16 have been developed to enhance their light-harvesting ability, charge mobility, self-assembling, solubility, and turning their molecular energy levels to meet the requirements of organic electronic devices.5,17–20 Poly(thienylene-vinylene)s (PTVs), which is one of polythiophene derivatives (PTs) containing alternating thiophene and vinylene units along the main-chains, have been intensively studied for their photoelectrical property and photovoltaic applications, due to their good charge transport property.2,21–25 In addition, previous literatures also have shown that polythiophene derivatives (PTs) with thienylene-vinylene conjugated side chains possess broader absorption spectra and relatively low-lying HOMO energy levels compared with their linear analogues, since the thienylene-vinylene conjugated side chains could extend the π-electron delocalization of polymer backbone.2,26–30 For example, chemically grafting conjugated side chains onto polymer backbone through vinylene-linkage, such as thienylene-vinylene31 and bi(thienylenevinylene),11 is effective to extend the absorption range and down-shift the HOMO energy level of polythiophene. However, the steric hindrance of bulky branches frequently lowers the coplanarity of thiophene rings on the main chain, leading to weak crystallinity and low π–π* absorption intensity.28 To overcome this problem, unsubstituted thienyl moieties have been inserted into the backbone as spacer to reduce the grafting density and increase the distance between conjugated branches.32 Nevertheless, the introduction of such electron-rich spacer is accompanied by the substantial decline of solubility and the raise of HOMO level, thus complicating the fabrication process and limiting the maximum attainable open-circuit voltage (Voc) of solar devices, respectively.Recently, we established a new class of polythiophene derivatives with terthiophene-vinylene (TTV) conjugated side chains, P1, whose structure is illustrated in Fig. 1, by chemically attaching polythiophene backbone with branched side chains through vinylene bridge.13 This polymer has adequate solubility, an extended absorption spectrum, a deep HOMO level and, more importantly, retains excellent crystallinity. It has been well documented that the bandgap reduction of polymer donor in organic photovoltaics is crucial in enhancing the photocurrent and then the power conversion efficiency (PCE). One important approach toward red-shifting the absorption band is the integration of electron-rich and electron-deficient units into the polymer backbone or conjugated side chains that would enhance effectively intramolecular charge transfer (ICT) to reduce the optical bandgap. In comparison to alternative copolymerization of these monomers, the strategy of random copolymerization has advantages of further broadening the absorption region caused by multiple chromophores.18,28,30,33 Overall, these strategies, such as incorporating electron-withdrawing groups into polymer backbone and/or conjugated side chains, would be conducive to absorption ability of conjugated polymer.
 | ||
| Fig. 1 Chemical structures of P1, P2, and P3 (R = 2-ethylhexyl; R′ = n-hexyl). | ||
Herein, we designed and synthesized two 2-D conjugated polymers, P2 and P3 (Fig. 1), by attaching two carbonyl groups to the TTV conjugated side chains and incorporating 2,1,3-benzothiadiazole (BTD) units into the polymer backbone. Because of the attachment of two carbonyl groups to the TTV conjugated side chains, P2 displays a deeper HOMO energy level and better solubility, and its absorption band from conjugated side chain is red-shifted in comparison to P1. In addition, BTD, a planar and strongly electron-withdrawing unit, is widely used to build high-performance donor–acceptor copolymers.34,35 Importantly, due to the randomized incorporation of steric-less BTD units into the polymer backbone of terpolymer, P3, its presence not only broadened the polymers absorption band but also promoted its self-assembling ability.36 The best PSC devices based on P3/PC61BM gave a PCE of 4.17% under AM1.5G illumination with an intensity of 100 mW cm−2. The effects of molecular structure design on the optical, electrical, crystallographic, and photovoltaic properties of these copolymers were systematically investigated.
Results and discussion
The synthetic routes used to prepare the new monomer, M2, and the target copolymers are shown in Scheme 1. On the basis of the reactivity of M1, it was anticipated that Friedel–Crafts acylation would preferentially occur at the 5 and 5′′ positions of the terthiophene side chain. Therefore, the new monomer M2 was synthesized using M1, heptanoyl chloride, and aluminum chloride. A coupling constant of ca. 16.0 Hz in the 1H NMR spectrum of M2 (Fig. S1 in the ESI†) clearly indicates that the E-isomer is the exclusive product. Detailed characterization data for this monomer is given in the ESI.†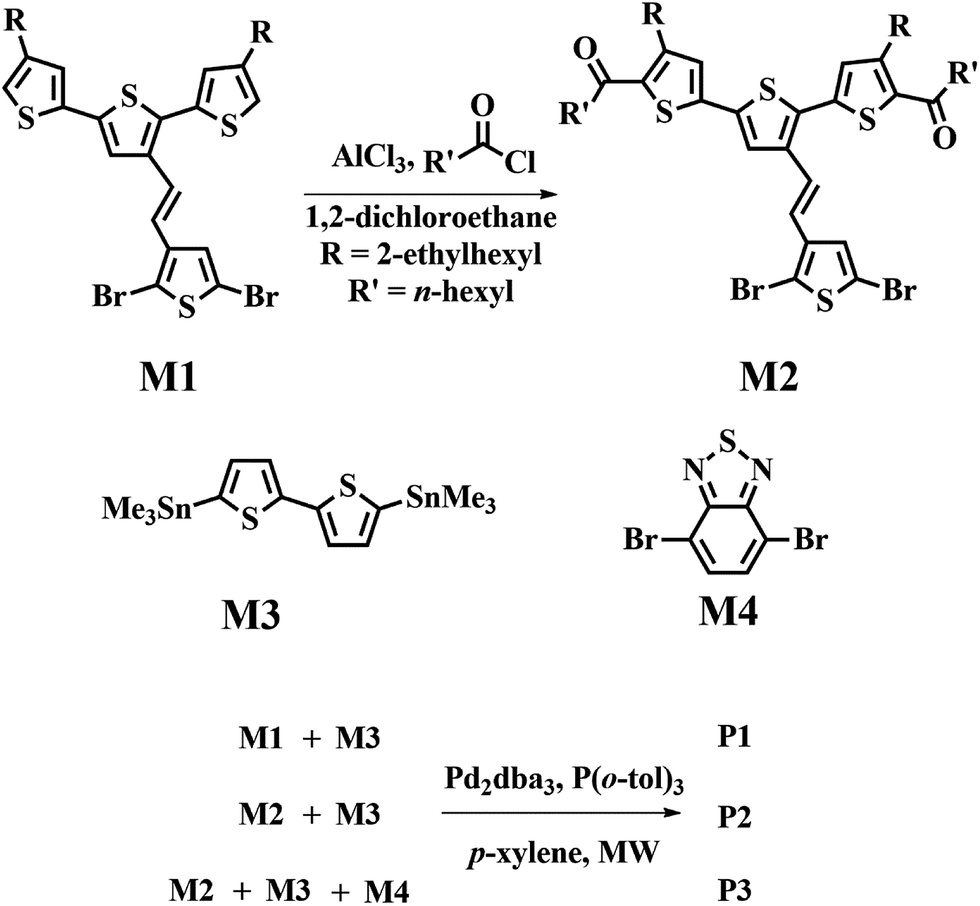 | ||
| Scheme 1 Synthetic routes for monomers and the corresponding polymers. | ||
All polymers were polymerized under microwave-assisted conditions. Microwave-assisted Stille polymerization lends itself very well to our polymerization, because conditions can be quickly optimized and the reaction time was dramatically shortened.37 We tried to simply optimize the polymerization conditions of P1 by varying the catalyst, monomer concentration, and microwave heating conditions. The results of the polymerization experiments are listed in Table S1 (ESI†). The results show that lower catalyst content, higher monomer concentration, and longer reaction time seem to produce polymers with higher molecular weights and lower polydispersity index (PDI) in polymerization of P1.
The optimized polymerization conditions of P1 were further applied in the synthesis of P2 and P3. Terpolymer P3 was polymerized with a molar feed ratio of M2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :M3:M4 of 2:3:1. It is noteworthy that the polymerization of random copolymer P3 is highly reproducible by microwave-assisted polymerization. The raw products were precipitated into methanol and collected through a Soxhlet thimble by filtration, which was then subjected to repeated Soxhlet extraction with methanol, acetone, hexane, and methylene chloride to remove the small molecules and oligomers, and finally with chloroform to collect the target compounds.
:M3:M4 of 2:3:1. It is noteworthy that the polymerization of random copolymer P3 is highly reproducible by microwave-assisted polymerization. The raw products were precipitated into methanol and collected through a Soxhlet thimble by filtration, which was then subjected to repeated Soxhlet extraction with methanol, acetone, hexane, and methylene chloride to remove the small molecules and oligomers, and finally with chloroform to collect the target compounds.
P1 and P2 have comparable yields of ca. 80% and exhibit good solubility in common solvents such as tetrahydrofuran (THF), chloroform, chlorobenzene (CB), and o-dichlorobenzene (oDCB). Moreover, gel permeation chromatography (GPC) shows that they have very similar molecular weight characteristics with number-average molecular weights of around 27 kDa with respect to polystyrene standards, as indicated in Table 1. However, P3 displays lower solubility in THF than the other polymers and therefore it is unavailable to obtain the molecular weight data by GPC. By using elemental analysis, the average m/n ratio in P3 was calculated to be 4.4, indicating that there is ca. 18% BTD units into the polymer backbone in reality.
| Polymer | Mna (kDa) | PDIa | Tdb (°C) | Solution, λmaxc (nm) | Film, λmaxd (nm) | Film, λonsetd (nm) | Eoptge (eV) | EHOMOf (eV) | ELUMOg (eV) |
|---|---|---|---|---|---|---|---|---|---|
| a Mn and PDI of the polymers were estimated by GPC using polystyrene as standards in THF.b 5% weight-loss temperature measured by TGA under N2 atmosphere.c Measured in dilute CB solution (10−3 g L−1).d Measured on glass substrates spin-coated with polymers from a CB solution (10 g L−1) followed by thermal annealing (120 °C, 15 min).e Estimated from the onset wavelength of the absorption spectra: Eoptg = 1240/λonset.f HOMO energy levels were evaluated by ultraviolet photoelectron spectroscopy (AC2 equipment) of thin polymer films on glass substrates.g Calculated according to the equation: ELUMO = EHOMO + Eoptg (eV). | |||||||||
| P1 | 27.0 | 3.94 | 449 | 341, 528 | 355/564 | 654 | 1.90 | −4.90 | −3.00 |
| P2 | 27.7 | 2.81 | 419 | 398, 510 | 406/557 | 669 | 1.85 | −5.14 | −3.29 |
| P3 | ✗ | ✗ | 430 | 410, 579 | 407/579 | 743 | 1.67 | −5.07 | −3.40 |
Thermal properties of all polymers were investigated by thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) analysis, as shown Fig. S7 in the ESI.† TGA plots reveal that the temperatures with 5% weight loss of P1, P2, and P3 are 449, 419, and 430 °C, respectively, under N2 atmosphere at a heating rate of 10 °C min−1. All compounds exhibit excellent thermal stability with degradation temperatures (Td) at 5% weight loss well beyond 400 °C. DSC plots were measured up to 300 °C, P1 exhibits an obvious phase transition at 282.1 °C which is considered its melting point. However, the other bulk samples display no apparent thermal transition.
UV-vis absorption spectra of the polymers in dilute CB solution and as spin-coated film on glass substrates are shown in Fig. 2. The corresponding absorption data of the polymers is summarized in Table 1. Comparison of the UV-vis spectra of P2 and P3 with control polymer P1 reveals how the structural modifications of the side and main chains affect the optical properties of branched-type 2-D PTs. First, in dilute CB solution, P2 displays two distinct absorption bands between 300 and 650 nm. The low-energy band that peaks at ∼535 nm corresponds to the π–π* transition of the conjugated backbone, whereas the high-energy (short wavelength) band that peaks at ∼398 nm and exhibits a distinct red-shift (of 57 nm) compared to P1 that originates from the intramolecular charge transfer between two electron-withdrawing heptanoyl groups and the terthiophene unit in the TTV conjugated side chain. However, owing to the replacement of H-atoms by two heptanoyl groups in the TTV conjugated side chain, the conjugated side chains become more bulky to disturb the coplanarity of the polymer backbone and therefore its π–π* transition peak is observed with a slight blue-shift (of 18 nm) and relatively weak intensity compared to P1. Furthermore, in comparison with P2, random copolymer P3 possesses a significantly broader absorption region covering a wavelength range of 300–750 nm. P3 also exhibits two distinct absorption bands at 407 nm and 579 nm. Because of the introduction of BTD units in the polymer backbone, the low-energy absorption band displays obvious red-shift that is mainly attributed to the ICT interaction of the BTD and BT units in the polymer backbone. Importantly, the partial bulky M2 units were replaced by less steric BTD units that would also enhance π–π* transition absorption due to reducing the twisting of polymer backbone. Through simple structural modifications of the side and main chains, we have successfully extended the absorption region of branched-type 2-D PTs, thereby enhancing their light-harvesting ability.
 | ||
| Fig. 2 UV-vis absorption spectra of polymers (a) in dilute CB solution at room temperature; (b) solid films prepared by spin-coating followed by thermal annealing at 120 °C for 15 min. | ||
As shown in Fig. 2(b), the polymer films generally display red-shifted and broader absorption than their corresponding solutions due to interchain associations and better planarity in the solid state.38 Therefore, the maximum values of the low-energy absorption peaks are red-shifted by ca. 36 nm, 47 nm, and 23 nm for the P1, P2, and P3 films, respectively, in comparison with solutions. Moreover, the optical bandgaps (Eoptg) of P1, P2, and P3, estimated from the absorption onsets of their solid films, are 1.90, 1.85, and 1.67 eV, respectively. P3 obviously displays a lower bandgap than P2 because of the incorporation of strong electron-withdrawing BTD units into the polymer backbone. The absorption edge of random terpolymer P3 is red-shifted to the near-infrared region, implying that it has great potential for effective photon-harvesting and could achieve a high short-circuit current in PSCs.27
Molecular structural design is important for organic materials that directly has effect on polymer orientation in thin films, leading to decide the performance of charge transport. However, it's rare to be studied in 2-D PTs' system. Herein, to further understand the influence of molecular structure designs on solid-state crystallinity and polymer self-assembly in TTV–PTs, the two-dimensional grazing incidence X-ray diffraction (2D-GIXRD) technique was used to measure the intermolecular d-spacing and average crystallite size in the drop-cast thin films of polymer with thermal annealing procedure for investigating the behaviors of molecular packing orientation of P1–P3, as shown in Fig. S4.† The intermolecular d-spacing parameter and average crystallite size were determined from the diffraction peaks using Bragg's law and Scherrer's equations.39
As extracted from the out-of-plane 2D-GIXRD profile shown in Fig. 3, P1 thin film possesses a visible (100) diffraction peak at 2θ = 3.01° (corresponding to an interlayer d-spacing of 2.54 nm with an average crystallite size of 16.5 nm), indicating lamellar packing parallel to the surface normal with an edge-on (100) orientation of the conjugated plane.40 As two hydrogen atoms were replaced by two heptanoyl substituents at 5 and 5′′ positions of the terthiophene side chain of P1, polymer P2 thin-film displayed amorphous nature that indicating the more bulky side groups in P2 hinder the molecular stacking. Therefore, although the additional two heptanoyl groups in TTV side chains could enhance the solubility and red-shift absorption in UV-vis region, it is destructive for polymer self-assembly and backbone coplanarity by larger side chains.
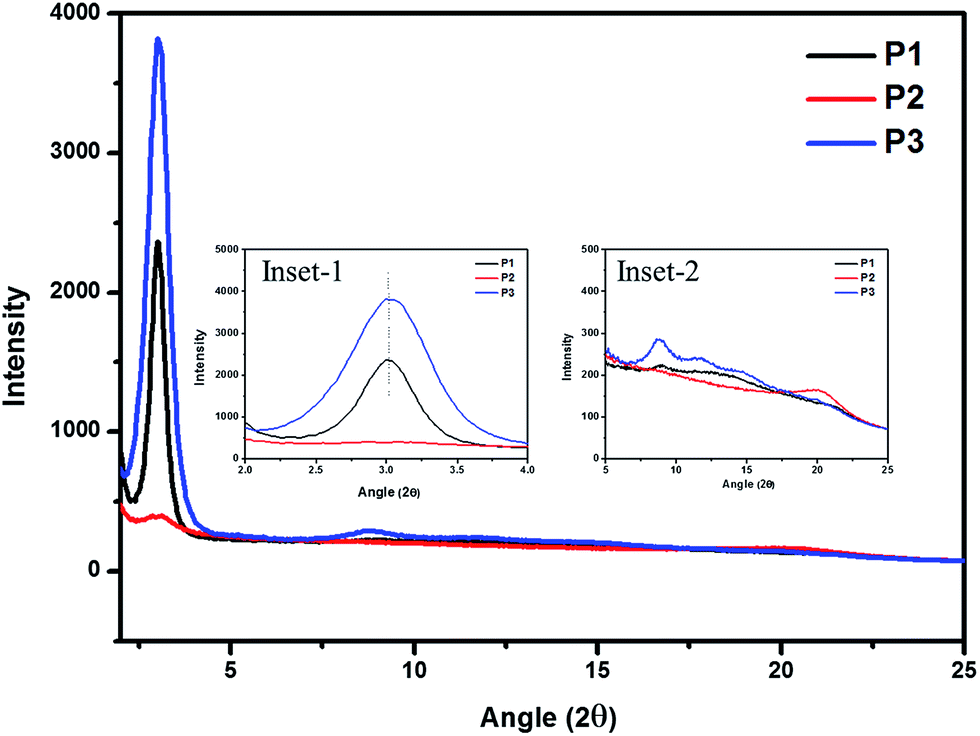 | ||
| Fig. 3 Grazing incidence X-ray diffraction patterns of polymer films prepared by drop-cast followed by thermal annealing at 120 °C for 15 min. The inset-1 and inset-2 shows the region around 2θ = 2–4° and 5–25°, respectively, in greater detail. | ||
Interestingly, P3 displays a strong (100) diffraction peak at 2θ = 3.01° (corresponding to a lamellar d-spacing of 2.54 nm with an average crystallite size of 11.3 nm). In other words, partial bulky M2 units were replaced by less sterically hindering BTD (M4) units in the polymer backbone that release enough free space to reduce the steric effect on the polymer backbone and promote molecular organization. This is an important strategy to induce polymer self-assembly for 2-D polythiophene derivatives by random copolymerization with less-steric units, such as 2,1,3-benzothiadiazole. In addition, compared to P1, the FWHM and intensity of (100) diffraction peak for P3 thin films is broader and stronger, respectively, indicating P3 thin films have higher degree of crystallinity and smaller average crystalline grain.
The charge mobility of conjugated polymers exerts a significant influence on the FF value of PSC devices,41 and many studies have revealed that the hole mobility of a crystalline conjugated polymer is usually higher than that of an amorphous conjugated polymer and therefore is more comparable with the electron mobility in the active layer, where the electron mobility is dominated by fullerene.42 The hole mobilities of polymers were measured in a hole-only device (ITO/PEDOT:PSS/pristine polymer/Au) using the space-charge-limited current (SCLC) method. Fig. S6(a)† which is plotted as ln(JdarkL3V−2) versus (VL−1)0.5 in ESI† shows the dependence of the measured current density on the applied voltage. The mobilities were calculated according to the following formula:43
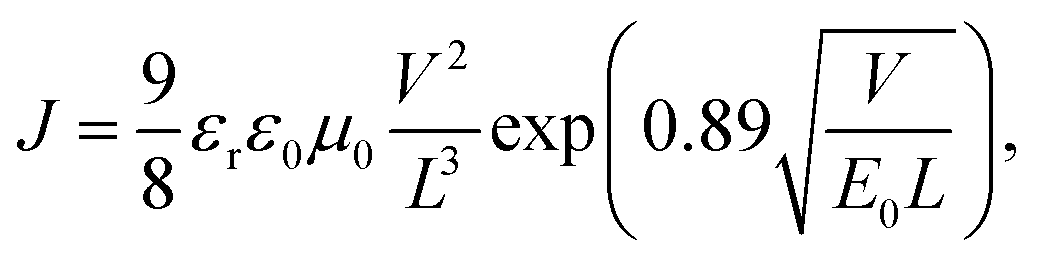
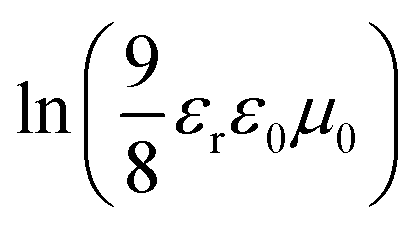 . Therefore, the hole mobility of pristine P1, P2, and P3 were determined to be 3.9 × 10−4 cm2 V−1 s−1, 3.8 × 10−5 cm2 V−1 s−1, and 1.0 × 10−4 cm2 V−1 s−1, respectively. It should be noted that thin film of P1 exhibited a diffraction peak at 2θ = 20.17°, shown in inset figure in Fig. 3, that would be classified as face-on orientation and correspond to a π–π stacking distance of 3.81 Å. Therefore, it is reasonable to explain that the pristine P1 thin film possesses the highest hole mobility by SCLC method, because it's well known that face-on polymer orientations have been suggested to promote the charge mobility by SCLC method and PSCs;44,45 meanwhile, the pristine P3 thin film takes an edge-on orientation nature on the substrate which might be caused by the bulky side groups hindering the development of π–π stacking of polymer backbone. Nevertheless, it is evident that the formation of an ordered polymer-chain structure improved the carrier mobility of the polymer material.
. Therefore, the hole mobility of pristine P1, P2, and P3 were determined to be 3.9 × 10−4 cm2 V−1 s−1, 3.8 × 10−5 cm2 V−1 s−1, and 1.0 × 10−4 cm2 V−1 s−1, respectively. It should be noted that thin film of P1 exhibited a diffraction peak at 2θ = 20.17°, shown in inset figure in Fig. 3, that would be classified as face-on orientation and correspond to a π–π stacking distance of 3.81 Å. Therefore, it is reasonable to explain that the pristine P1 thin film possesses the highest hole mobility by SCLC method, because it's well known that face-on polymer orientations have been suggested to promote the charge mobility by SCLC method and PSCs;44,45 meanwhile, the pristine P3 thin film takes an edge-on orientation nature on the substrate which might be caused by the bulky side groups hindering the development of π–π stacking of polymer backbone. Nevertheless, it is evident that the formation of an ordered polymer-chain structure improved the carrier mobility of the polymer material.
The highest occupied molecular orbital (HOMO) energies were estimated from the ionization potentials of spin-coated films determined by UV photoelectron spectroscopy in air using an AC2 photoelectron spectrometer (Riken Keiki Co.).46 Fig. S3† plots the square root of the counting rate (CR) as a function of the photon energy. The photoemission threshold energy, also called the work function, was determined from the crossing point of the background and yield lines. Therefore, the work functions of P1, P2, and P3, were found to be −4.90 eV, −5.14 eV, and −5.07 eV, respectively (see Table 1) and the energy-level diagrams of polymers derived from AC2 photoelectron spectroscopy and UV-vis absorption data are shown in Fig. 4. Furthermore, the lowest unoccupied molecular orbital (LUMO) energies were calculated using the optical band gap (Eoptg) and the HOMO energies determined from AC2 measurement according to the following equation: ELUMO = EHOMO + Eoptg (eV).
 | ||
| Fig. 4 Energy diagram of P1, P2, and P3. | ||
By comparing with the HOMO of P1, the HOMO of P2 is 0.24 eV deeper. Because two additional electron-withdrawing heptanoyl substituents attached to the 5 and 5′′ positions of the terthiophene side chain that enhance its air stability in ambient conditions, P2 possessed further down-shifted HOMO level than P1. Furthermore, M2 units were partially replaced by M4 units in the backbone of P3 such that the lower ratio of monomers containing conjugated side chains in the polymer backbone resulted in the HOMO rising. Similar results have also been observed for other 2-D PTs random copolymers.27,29,47 In addition, the differences (>0.5 eV) in the LUMO levels of the polymers and PC61BM (∼4.20 eV) are still adequate enough for efficient charge transfer, even for the lowest LUMO in P3.27,48 The AC2 measurements reveal that our series of 2-D conjugated polymers have lower HOMO energies than rr-P3HT (ca. −4.68 eV under identical condition); this will be beneficial for the fabrication of photovoltaic devices with high Voc values.
The broader absorption bands and lower-lying HOMO energy levels of our polymers suggest that they might exhibit promising photovoltaic properties. Bulk heterojunction PSC devices were fabricated using P1, P2, or P3 as the electron donor, and PC61BM as the electron acceptor. The device structure is ITO/PEDOT:PSS/polymer:PC61BM/Ca/Al. The cell performance was optimized by varying the polymer to PC61BM blend ratio in the active layer and thermal annealing temperature. The J–V curves of the optimized polymer/fullerene devices under AM1.5G illumination with an intensity of 100 mW cm−2 are plotted in Fig. 5(a) and the relevant photovoltaic characteristics are listed in Table 2. The highest PCEs (%) for each PSC device are 3.89% (P1/PC61BM 1:0.8 w/w), 1.52% (P2/PC61BM 1:0.8 w/w), and 4.17% (P3/PC61BM 1:1 w/w).
 | ||
| Fig. 5 (a) J–V curves (b) IPCE spectra of optimized polymer/PC61BM devices under AM1.5G illumination with an intensity of 100 mW cm−2. | ||
| Polymer/fullerene | Voc (mV) | Jsc (mA cm−2) | FF (%) | PCE (%) | Rsh (kΩ cm2) | Rs (Ω cm2) |
|---|---|---|---|---|---|---|
| a Blend ratio 10:8 (w/w), in CB solvent, annealing at 100 °C for 10 min.b Blend ratio 10:8 (w/w), in o-DCB solvent, annealing at 80 °C for 10 min.c Blend ratio 7:7 (w/w), in o-DCB solvent, annealing at 120 °C for 10 min. |
||||||
| P1/PC61BMa | 788 ± 4 | 7.24 ± 0.05 | 67.76 ± 0.27 | 3.87 ± 0.02 (3.89) | 1.01 ± 0.03 | 9.03 ± 0.27 |
| P2/PC61BMb | 688 ± 4 | 5.50 ± 0.28 | 38.25 ± 0.44 | 1.44 ± 0.08 (1.52) | 0.47 ± 0.03 | 58.94 ± 2.75 |
| P3/PC61BMc | 770 ± 0 | 9.25 ± 0.13 | 57.59 ± 0.63 | 4.10 ± 0.07 (4.17) | 0.86 ± 0.09 | 11.61 ± 0.52 |
The incident photo-to-electron conversion efficiencies (IPCE) of the optimized polymer/fullerene devices under AM1.5G illumination with an intensity of 100 mW cm−2 are plotted in Fig. 5(b). The Jsc values calculated by integrating the IPCE data with the AM1.5G spectrum were 7.64, 5.86 and 9.55 mA cm−2 for P1, P2 and P3 devices, respectively. The IPCE results not only agree with the measured Jsc values, considering that an error of 3–6% commonly takes place for the IPCE measurements, but also exhibit well-matched curves with their optical absorptions.27,30,49 The IPCE spectra of the P1/PC61BM (1:0.8 w/w) device covers a wavelength range of 350–650 nm. Moreover, the device based on P2/PC61BM and P3/PC61BM show higher IPCE values in the wavelength range of 400–430 nm and 370–475 nm, respectively, than P1/PC61BM. This result reveals that two additional heptanoyl substituents attached to the TTV conjugated side chain can effectively enhance absorption bands between 350 and 480 nm as shown in Fig. 2(b) and further lead to increase photocurrent generation in the region. However, the P2/PC61BM device shows a rather low IPCE over most of the response range, thus causing its lower Jsc and PCE. For the P3/PC61BM device, the active region of the IPCE curve covers the broadest wavelength range from 300 to 750 nm. The existence of the two heptanoyl substituents in P3 promote the higher IPCE values from 370 to 475 nm compared to the P1 device; meanwhile, by incorporating BTD units into the P3 backbone, the IPCE curve is further extended to 750 nm, thus causing this device to have the broadest IPCE region and highest Jsc.
The electron and hole mobilities of the blended films were determined by using the SCLC method to gain an insight into their effective charge carrier mobilities in the optimized blend weight ratio. Experimental results shown in Fig. S6(b) and (c) and Table S2† indicate that the trend in hole mobility of the blended films is the same as that for the series resistance (Rs). As a result, the hole mobility of P2/PC61BM is approximately two orders of magnitude lower than the electron mobility, which leads to unbalanced charge carrier transport and recombination, and may be responsible for the poor photovoltaic characteristics observed.
The open-circuit voltage (Voc) of PSC device is mainly related to the difference between the HOMO of the donor and LUMO of the acceptor.48 Thus, because of the low-lying HOMO energy levels of PTs containing conjugated side chains, an average Voc of 788 mV was obtained from the P1/PC61BM devices, which substantially exceeds the commonly reported Voc (∼0.6 V) for devices based on P3HT/PC61BM.50–52 However, the lowest Voc value was observed for the P2/PC61BM device which is quite poorly consistent with the position of the P2 HOMO energy level. The non-linear trend is decided by a couple of other factors besides the HOMO levels of the polymers, such as the dark current, blend morphology of the photoactive layers, and interface resistance.30,34,53–55 Fig. S5 (shown in ESI†) shows dark J–V curves of polymer/PC61BM devices. The dark current of the P2/PC61BM device was obviously larger than that of the P1/PC61BM device implying that serious recombination from dark carriers might have caused a loss of open-circuit voltage.
The morphology of photoactive layer plays a decisive role in determining the photovoltaic characteristics of PSC devices.56–58 Herein, atomic force microscopy (AFM) was used to investigate the morphologies of the spin-coated films of polymers/PC61BM on top of ITO/PEDOT:PSS, which were prepared by using the same procedure and parameters for preparing the photoactive layers. As shown in Fig. 6, the P2/PC61BM blend film exhibits the smoothest surface and the least degree of phase separation with a root-mean-square (RMS) roughness of 2.16 nm and a phase degree of 0.21°. However, P2/PC61BM has the slowest hole mobility of 1.1 × 10−6 cm2 V−1 s−1, which is also only around one-third of that of pristine P2. These findings suggest that the polymer chains of P2 form homogeneous mixture with PC61BM and are unable to effectively segregate into continuous and unhindered pathways for conduction of holes to the anode. Therefore, the Jsc of the P2/PC61BM solar cell is much smaller than that of both P1/PC61BM and P3/PC61BM devices.
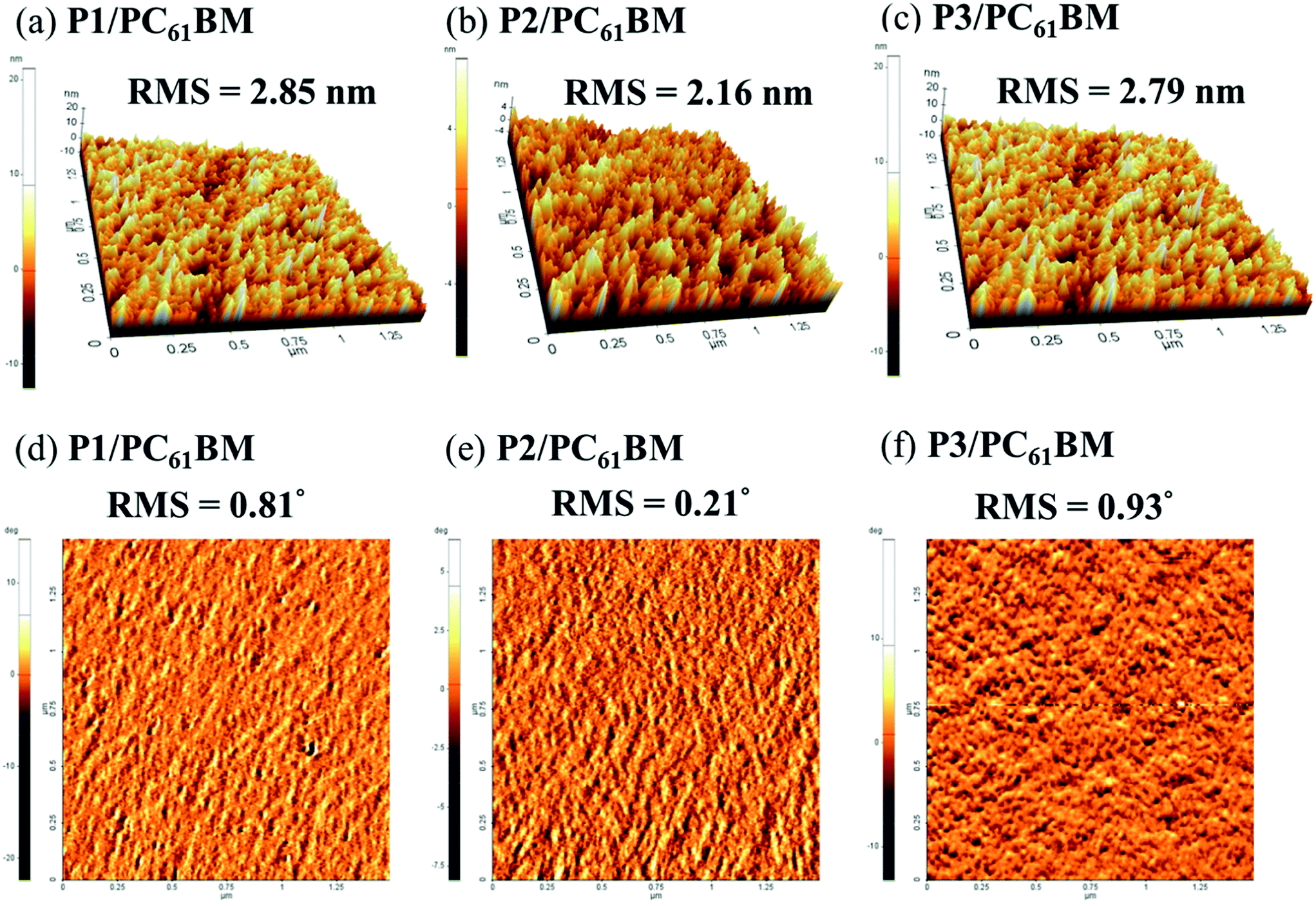 | ||
| Fig. 6 AFM (a–c) topography images and (d–f) phase images of spin-coated films of polymer/PC61BM blends. The scan sizes for all images are 2 μm × 2 μm. | ||
Conclusions
We have successfully incorporated electron-withdrawing units, heptanoyl group and BTD moiety, into the TTV conjugated side chains and polythiophene backbone to synthesize TTV–PTs, P2 and P3. Compared with their parent analogue, these polymers possess apparently red-shifted absorption bands as well as down-shifted HOMO and LUMO levels that are beneficial to improve the polymer donor's light-harvesting ability and lift the Voc of solar cells. However, the steric hindrance of bulky heptanoyl groups in P2 significantly twists the polymer backbone, not only shortening the conjugation length but also lowering the crystallinity and then hole mobility. It is interesting to note that the crowding effect of oversized side groups can be completely relaxed by the insertion of 18% BTD units into the polymer main chain through a simple random copolymerization method and, therefore, the resulting terpolymer P3 has broad absorption band, excellent crystallinity and good carrier mobility. Consequently, the bulk heterojunction solar cell fabricated from P3/PC61BM exhibits a promised PCE of 4.17% with a Voc of 770 mV, a Jsc of 9.3 mA cm−2 and an FF of 58%. A further improvement can be made by introducing larger solubilizing group onto the TTV unit to allow more BTD to be merged into the backbone. More importantly, this study offers an easy and useful strategy to design highly crystalline 2D conjugated polymers for developing high-performance polymer solar cells.Experimental section
Materials
(E)-3′-(2-(2,5-Dibromothiophen-3-yl)vinyl)-4,4′′-bis(2-ethylhexyl)-2,2′:5′,2′′-terthiophene (M1),59 5,5′-bis(trimethylstannyl)-2,2′-bithiophene (M3),60 4,7-dibromo-2,1,3-benzothiadiazole (M4)61 were prepared via literature procedures. Toluene (Acros Organics, 99+%, extra pure) and p-xylene (Acros Organics, 99+%, extra pure) and methylene chloride (ECHO Chemical, ACS Grade, 99.5%) are dried over calcium hydride (Acros Organics, ca. 93%, extra pure, 10–100 mm pieces) under N2 and stored with molecular sieves in a round-bottom flask under inert atmosphere. Flash chromatography was performed over silica gel (230–400 mesh).(E)-1,1′-(3′-(2-(2,5-Dibromothiophen-3-yl)vinyl)-4,4′′-bis(2-ethylhexyl)-[2,2′:5′,2′′-terthiophene]-5,5′′-diyl)bis(heptan-1-one) (M2)
A solution of AlCl3 (Acros Organics, 99%, 346.7 mg, 2.6 mmol) in anhydrous 1,2-dichloroethane (50 mL) was added to a 100 mL Schlenk flask under N2 atmosphere. Heptanoyl chloride (Alfa Aesar, 99%, 0.4 mL, 2.6 mmol) freshly distilled was then slowly added dropwise to the solution. The mixture was stirred at room temperature for 30 min and then cooled to 0 °C. Subsequently, M1 (960 mg, 1.3 mmol) in anhydrous 1,2-dichloroethane (10 mL) was slowly added dropwise to the solution, causing the color of the suspension to change from light yellow to deep-red. The mixture was warmed to room temperature and stirred overnight. Then 2 M hydrochloric acid (100 mL) was slowly added to quench the reaction and the resulting mixture were extracted twice with 40 mL dichloromethane, then the organic phase was combined, washed several times with saturated sodium acetate solution and water, and dried with MgSO4. After removal of the solvent, the residue was purified by flash column chromatography (eluent: hexane/CH2Cl2, 1:1, v/v), yielding a pale-yellow oil M2 (601.0 mg, 0.62 mmol, 48%). 1H NMR (400 MHz, CDCl3) δ 7.43 (s, 1H), 7.07 (d, 1H, J = 16 Hz), 7.07 (s, 1H), 7.03 (s, 1H), 6.98 (s, 1H), 6.87 (d, 1H, J = 16 Hz), 2.99–2.88 (m, 4H), 2.81–2.76 (m, 4H), 1.76–1.65 (m, 6H), 1.35–1.26 (m, 28H), 0.90–0.87 (m, 18H); 13C NMR (100 MHz, CDCl3) δ 193.26, 193.21, 150.65, 150.34, 138.72, 138.57, 137.24, 137.11, 136.19, 134.78, 132.43, 131.85, 129.08, 127.13, 123.54, 123.33, 122.76, 112.32, 110.87, 42.40, 42.30, 40.19, 34.50, 32.66, 32.55, 31.60, 28.95, 28.68, 28.62, 25.74, 24.76, 24.70, 23.06, 22.49, 14.08, 14.01, 10.71 ppm; HRMS (ESI+): m/z calcd for C48H67O2Br2S4: 963.2365 [M+]; found: 963.2365.
Microwave-assisted polymerization procedure
Monomers were weighted into microwave tubes (G10) and then the tubes were subjected to three successive cycles of vacuum followed by refilling with nitrogen. After addition of Pd-catalyst (Alfa Aesar, tris(dibenzylideneacetone)dipalladium(0), [Pd2(dba)3], Pd 21.5% min; Acros Organics, tri-o-tolylphosphine, 99%; Acros Organics, tetrakis(triphenylphosphine)palladium(0), [Pd(PPh3)4], 99%) in toluene or p-xylene solution (3 mol% relative to 5,5′-bis(trimethylstannyl)-2,2′-bithiophene, M3), the tubes were subjected to the reaction conditions in a microwave reactor. The heating condition: raise temperature from r.t. to 200 °C as fast as possible; hold the temperature 60 min; cool down to 55 °C. Each raw product was dissolved in toluene, then precipitated into methanol and collected through a Soxhlet thimble by filtration and then subjected to consecutive Soxhlet extractions with methanol, acetone, hexane, and methylene chloride to remove the small molecules and oligomers, and finally with chloroform to collect the target compounds.P1 is synthesized according to the procedure with M1 (182.5 mg, 0.247 mmol) and M3 (122.3 mg, 0.249 mmol) in 5.0 mL p-xylene, yielding P1 (154.6 mg, 84.1%).
P2 is synthesized according to the procedure with M2 (167.4 mg, 0.174 mmol) and M3 (86.1 g, 0.175 mmol) in 3.5 mL p-xylene, yielding P2 (136.5 mg, 81.0%).
P3 is synthesized according to the procedure with M2 (203.6 mg, 0.211 mmol), M3 (156.0 mg, 0.317 mmol) and M4 (31.1 mg, 0.105 mmol) in 6.3 mL p-xylene, yielding P3 (104.5 mg, 49.5%). Elemental analysis: found (average value): S, 20.620%; H, 7.221%; C, 68.015%; N, 0.610%.
Characterization and instrumentation
Microwave assisted polymerizations were performed in Anton Paar Monowave 300 microwave reactor. 1H NMR and 13C NMR spectra were recorded at 400 MHz on Bruker DRX-400 spectrometer. All NMR spectra were calibrated by chloroform-d (CDCl3) where 1H NMR chemical shifts of CDCl3 is 7.23 ppm; in addition, 13C NMR of CDCl3 is 77.0 ppm. Microwave assisted polymerizations were performed in Anton Paar Monowave 300 microwave reactor. Thermal gravimetric analysis (TGA) was measured on TA Instruments Hi-Res TGA 2950 and differential scanning calorimetry (DSC) was recorded on PerkinElmer DSC 8000 under N2 atmosphere at a heating rate of 10 °C min−1. Ultraviolet-visible absorption (UV-vis) spectra were recorded on a JASCO MD-2010 spectrometer. Gel permeation chromatography (GPC) was conducted at 40 °C using two Jordi DVB mixed-bed columns (250 × 10 mm; suitable for separating polymers with molecular weights from 3 × 103 to 5 × 106 g mol−1) using THF as the eluent at a flow rate of 1.0 mL min−1 on a JASCO instrument that was equipped with UV-vis and refractive index (RI) detectors connected in series. 2D-GIXRD measurements were carried out on the wiggler beamline BL17A1 of the National Synchrotron Radiation Research Center (NSRRC, Taiwan), with a wavelength of 1.333 Å delivered from a superconducting wavelength-shifting magnet and a Si(111) triangular crystal monochromator (TCM). The grazing angle was large enough to probe the interior of the films and meantime small enough to increase the X-ray path length within the films by orders of magnitude.62 The data were recorded by a Mar3450 image plate with exposure duration 60 second. The two-dimensional diffraction pattern was converted to a one dimensional powder diffraction profile by fit-2D program. The diffraction patterns are fitted in the one dimensional powder diffraction spectra using Gauss function of OriginPro 8 to locate the peak positions and calculate the full-width at half-maximum. The work functions of materials were measured using an AC2 photoelectron spectrometer (Riken Keiki Co.). Atomic force microscopy (AFM) images were captured using taping model in Digital Instruments Nanoscope III. The AFM samples were prepared by spin-coating the blend solution on a ITO/PEDOT:PSS substrate.Device fabrication
The polymer solar cells (PSCs) were fabricated with a configuration of ITO/PEDOT:PSS/active layer/Ca/Al. The ITO glasses were cleaned by a sequential ultrasonic treatment in detergent, deionized water, acetone, and isopropanol for 20 min. Then PEDOT:PSS was filtered through a 0.2 μm filter and spin-coated at 3500 rpm for 30 s on top of ITO electrode. Subsequently, the PEDOT:PSS film was baked at 140 °C for 10 min in the air, and then moved into a glovebox. The blend solution of PC61BM and synthesized polymers in a solvent was filtered with/without a 0.45 μm filter and spin-coated at 800 rpm for 30 s on top of the PEDOT:PSS layer. These devices were thermally annealed at various temperatures for 10 min, followed by capping with Ca (∼20 nm) and then Al (∼60 nm) in a thermal evaporator at a base pressure of ca. 10−6 Pa. The active area of the devices is 0.06 cm2. The current density–voltage measurements of the devices were conducted on a computer-controlled Keithley 2400 Source Measure Unit under AM1.5G simulated solar irradiation at 100 mW cm−2. The light incident intensity was calibrated by a mono-Si reference cell with a KG5 filter (PV Measurements, Inc.), which was pre-calibrated by the National Renewable Energy Laboratory. The SCLC measurements were carried out using a Keithley 2400 source meter under dark condition. The IPCE spectra were recorded under illumination by a xenon lamp and a monochromator (TRIAX 180, JOBIN YVON), and the light intensity was calibrated by using an OPHIR 2A-SH thermopile detector.Acknowledgements
This research was financially supported by National Taiwan University, Academia Sinica (AS-103-SS-A02), and the Ministry of Science and Technology of the Republic of China (MOST 102-2113-M-002-003-MY3; MOST 105-3113-E-102-010). The authors thank the instrumentation center sponsored by Ministry of Science and Technology and National Taiwan University for NMR, elemental analysis and TEM experiments.References
- J. Mei and Z. Bao, Chem. Mater., 2014, 26, 604–615 CrossRef CAS.
- C. Lu, H. C. Wu, Y. C. Chiu, W. Y. Lee and W. C. Chen, Macromolecules, 2012, 45, 3047–3056 CrossRef CAS.
- P. M. Beaujuge and J. R. Reynolds, Chem. Rev., 2010, 110, 268–320 CrossRef CAS PubMed.
- A. C. Grimsdale, K. Leok Chan, R. E. Martin, P. G. Jokisz and A. B. Holmes, Chem. Rev., 2009, 109, 897–1091 CrossRef CAS PubMed.
- J. Mei, Y. Diao, A. L. Appleton, L. Fang and Z. Bao, J. Am. Chem. Soc., 2013, 135, 6724–6746 CrossRef CAS PubMed.
- K. Pu, A. J. Shuhendler, J. V. Jokerst, J. Mei, S. S. Gambhir, Z. Bao and J. Rao, Nat. Nanotechnol., 2014, 9, 233–239 CrossRef CAS PubMed.
- L. Feng, C. Zhu, H. Yuan, L. Liu, F. Lv and S. Wang, Chem. Soc. Rev., 2013, 42, 6620–6633 RSC.
- Z.-G. Zhang and J. Wang, J. Mater. Chem., 2012, 22, 4178–4187 RSC.
- P.-C. Jwo, Y.-Y. Lai, C.-E. Tsai, Y.-Y. Lai, W.-W. Liang, C.-S. Hsu and Y.-J. Cheng, Macromolecules, 2014, 47, 7386–7396 CrossRef CAS.
- J. Lee, S. B. Jo, M. Kim, H. G. Kim, J. Shin, H. Kim and K. Cho, Adv. Mater., 2014, 26, 6662 CrossRef CAS.
- J. Hou, Z. A. Tan, Y. Yan, Y. He, C. Yang and Y. Li, J. Am. Chem. Soc., 2006, 128, 4911–4916 CrossRef CAS PubMed.
- R. Duan, L. Ye, X. Guo, Y. Huang, P. Wang, S. Zhang, J. Zhang, L. Huo and J. Hou, Macromolecules, 2012, 45, 3032–3038 CrossRef CAS.
- C.-Y. Hsiow, R. Raja, C.-Y. Wang, Y.-H. Lin, Y.-W. Yang, Y.-J. Hsieh, S.-P. Rwei, W.-Y. Chiu, C.-I. Huang and L. Wang, Phys. Chem. Chem. Phys., 2014, 16, 25111–25120 RSC.
- X. Liu, Y. Huang, Z. Cao, C. Weng, H. Chen and S. Tan, Polym. Chem., 2013, 4, 4737–4745 RSC.
- C. Wang, B. Zhao, Z. Cao, P. Shen, Z. Tan, X. Li and S. Tan, Chem. Commun., 2013, 49, 3857–3859 RSC.
- C.-Y. Kuo, W. Nie, H. Tsai, H.-J. Yen, A. D. Mohite, G. Gupta, A. M. Dattelbaum, D. J. William, K. C. Cha, Y. Yang, L. Wang and H.-L. Wang, Macromolecules, 2014, 47, 1008–1020 CrossRef CAS.
- P.-L. T. Boudreault, A. Najari and M. Leclerc, Chem. Mater., 2010, 23, 456–469 CrossRef.
- Y. Li, Acc. Chem. Res., 2012, 45, 723–733 CrossRef CAS PubMed.
- S. Zhang, Z.-G. Zhang and Y. Li, Adv. Polym. Technol., 2013, 32, E822–E831 CrossRef CAS.
- X. Meng, W. Zhang, Z. A. Tan, C. Du, C. Li, Z. Bo, Y. Li, X. Yang, M. Zhen, F. Jiang, J. Zheng, T. Wang, L. Jiang, C. Shu and C. Wang, Chem. Commun., 2012, 48, 425–427 RSC.
- K.-Y. Jen, M. Maxifield, L. W. Shacklette and R. L. Elsenbaumer, J. Chem. Soc., Chem. Commun., 1987, 309–311 RSC.
- F. Goldoni, R. A. J. Janssen and E. W. Meijer, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 4629–4639 CrossRef CAS.
- J. Hou, Z. A. Tan, Y. He, C. Yang and Y. Li, Macromolecules, 2006, 39, 4657–4662 CrossRef CAS.
- Y. Qin and M. A. Hillmyer, Macromolecules, 2009, 42, 6429–6432 CrossRef CAS.
- Z. Zhang and Y. Qin, Macromolecules, 2016, 49, 3318–3327 CrossRef CAS.
- C.-J. Lin, W.-Y. Lee, C. Lu, H.-W. Lin and W.-C. Chen, Macromolecules, 2011, 44, 9565–9573 CrossRef CAS.
- Y. Liu, C.-C. Chen, Z. Hong, J. Gao, Y. Yang, H. Zhou, L. Dou, G. Li and Y. Yang, Sci. Rep., 2013, 3, 3356 CrossRef PubMed.
- M. Xu, M. Zhang, M. Pastore, R. Li, F. De Angelis and P. Wang, Chem. Sci., 2012, 3, 976–983 RSC.
- J. Hou, L. Huo, C. He, C. Yang and Y. Li, Macromolecules, 2006, 39, 594–603 CrossRef CAS.
- M. Zhang, F. Wu, Z. Cao, T. Shen, H. Chen, X. Li and S. Tan, Polym. Chem., 2014, 5, 4054–4060 RSC.
- J. Hou, C. Yang, C. He and Y. Li, Chem. Commun., 2006, 871–873 RSC.
- H. Tan, X. Deng, J. Yu, B. Zhao, Y. Wang, Y. Liu, W. Zhu, H. Wu and Y. Cao, Macromolecules, 2012, 46, 113–118 CrossRef.
- B. Burkhart, P. P. Khlyabich, T. Cakir Canak, T. W. LaJoie and B. C. Thompson, Macromolecules, 2011, 44, 1242–1246 CrossRef CAS.
- Z. He, C. Zhong, X. Huang, W.-Y. Wong, H. Wu, L. Chen, S. Su and Y. Cao, Adv. Mater., 2011, 23, 4636–4643 CrossRef CAS PubMed.
- R. C. Coffin, J. Peet, J. Rogers and G. C. Bazan, Nat. Chem., 2009, 1, 657–661 CrossRef CAS PubMed.
- P. P. Khlyabich, B. Burkhart, C. F. Ng and B. C. Thompson, Macromolecules, 2011, 44, 5079–5084 CrossRef CAS.
- B. Carsten, F. He, H. J. Son, T. Xu and L. Yu, Chem. Rev., 2011, 111, 1493–1528 CrossRef CAS PubMed.
- Z. Zhu, D. Waller, R. Gaudiana, M. Morana, D. Mühlbacher, M. Scharber and C. Brabec, Macromolecules, 2007, 40, 1981–1986 CrossRef CAS.
- U. Holzwarth and N. Gibson, Nat. Nanotechnol., 2011, 6, 534 CrossRef CAS PubMed.
- X. Zhang, L. J. Richter, D. M. DeLongchamp, R. J. Kline, M. R. Hammond, I. McCulloch, M. Heeney, R. S. Ashraf, J. N. Smith, T. D. Anthopoulos, B. Schroeder, Y. H. Geerts, D. A. Fischer and M. F. Toney, J. Am. Chem. Soc., 2011, 133, 15073–15084 CrossRef CAS PubMed.
- H.-L. Yip and A. K. Y. Jen, Energy Environ. Sci., 2012, 5, 5994–6011 Search PubMed.
- J.-M. Jiang, M.-C. Yuan, K. Dinakaran, A. Hariharan and K.-H. Wei, J. Mater. Chem. A, 2013, 1, 4415–4422 RSC.
- Y.-M. Chang, W.-F. Su and L. Wang, Sol. Energy Mater. Sol. Cells, 2008, 92, 761–765 CrossRef CAS.
- C. Piliego, T. W. Holcombe, J. D. Douglas, C. H. Woo, P. M. Beaujuge and J. M. J. Fréchet, J. Am. Chem. Soc., 2010, 132, 7595–7597 CrossRef CAS PubMed.
- J. W. Jo, J. W. Jung, E. H. Jung, H. Ahn, T. J. Shin and W. H. Jo, Energy Environ. Sci., 2015, 8, 2427–2434 Search PubMed.
- Y.-M. Chang, L. Wang and W.-F. Su, Org. Electron., 2008, 9, 968–973 CrossRef CAS.
- M.-C. Yuan, M.-Y. Chiu, C.-M. Chiang and K.-H. Wei, Macromolecules, 2010, 43, 6270–6277 CrossRef CAS.
- Y.-J. Cheng, S.-H. Yang and C.-S. Hsu, Chem. Rev., 2009, 109, 5868–5923 CrossRef CAS PubMed.
- B. C. Thompson and J. M. J. Fréchet, Angew. Chem., Int. Ed., 2008, 47, 58–77 CrossRef CAS PubMed.
- H. J. Son, F. He, B. Carsten and L. Yu, J. Mater. Chem., 2011, 21, 18934–18945 RSC.
- H.-W. Liu, D.-Y. Chang, W.-Y. Chiu, S.-P. Rwei and L. Wang, J. Mater. Chem., 2012, 22, 15586–15591 RSC.
- P. P. Khlyabich, B. Burkhart, A. E. Rudenko and B. C. Thompson, Polymer, 2013, 54, 5267–5298 CrossRef CAS.
- X. Chen, B. Liu, Y. Zou, W. Tang, Y. Li and D. Xiao, RSC Adv., 2012, 2, 7439–7448 RSC.
- S. Günes, H. Neugebauer and N. S. Sariciftci, Chem. Rev., 2007, 107, 1324–1338 CrossRef PubMed.
- N. Li, B. E. Lassiter, R. R. Lunt, G. Wei and S. R. Forrest, Appl. Phys. Lett., 2009, 94, 023307 CrossRef.
- F. Liu, Y. Gu, X. Shen, S. Ferdous, H.-W. Wang and T. P. Russell, Prog. Polym. Sci., 2013, 38, 1990–2052 CrossRef CAS.
- J. Wang, M. Xiao, W. Chen, M. Qiu, Z. Du, W. Zhu, S. Wen, N. Wang and R. Yang, Macromolecules, 2014, 47, 7823–7830 CrossRef CAS.
- J.-M. Jiang, P. Raghunath, H.-K. Lin, Y.-C. Lin, M. C. Lin and K.-H. Wei, Macromolecules, 2014, 47, 7070–7080 CrossRef CAS.
- C.-Y. Kuo, Y.-C. Huang, C.-Y. Hsiow, Y.-W. Yang, C.-I. Huang, S.-P. Rwei, H.-L. Wang and L. Wang, Macromolecules, 2013, 46, 5985–5997 CrossRef CAS.
- A. F. M. Kilbinger and W. J. Feast, J. Mater. Chem., 2000, 10, 1777–1784 RSC.
- R. S. Kularatne, F. J. Taenzler, H. D. Magurudeniya, J. Du, J. W. Murphy, E. E. Sheina, B. E. Gnade, M. C. Biewer and M. C. Stefan, J. Mater. Chem. A, 2013, 1, 15535–15543 RSC.
- D. M. Robinson, Y. B. Go, M. Greenblatt and G. C. Dismukes, J. Am. Chem. Soc., 2010, 132, 11467–11469 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra11738d |
| This journal is © The Royal Society of Chemistry 2016 |