DOI:
10.1039/C6RA11720A
(Paper)
RSC Adv., 2016,
6, 70791-70799
Microhydration effects on the structures and electrophilic properties of cytidine†
Received
5th May 2016
, Accepted 20th July 2016
First published on 20th July 2016
Abstract
Microhydration effects on the geometrical structures, electron affinities and charge distributions of cytidine and their anions have been investigated systematically using density functional theory (DFT), by explicitly considering cytidine complexes with up to four water molecules. Various structures of neutral and anionic cytidine(H2O)n (n = 2–4) have been predicted, and N3, H–N4 and O2 are found to be the most favorable water-binding sites of cytidine. The adiabatic electron affinities of cytidine(H2O)n increase linearly with the number of hydrating water molecules, indicating that they would obtain a stronger ability to attract electrons with the hydration number increasing. By examining the SOMO and natural population analysis, we found the excess electron density is localized on the cytidine moiety, especially on the cytosine base unit. This may help explain why the hydrogen bond changes upon the extra electron attachment. In addition, the maps of the reduced density gradient isosurfaces show a rich visualization of the hydrogen bond, van der Waals interaction and steric effect.
1. Introduction
In 2000, Sanche et al.1–3 demonstrated that low-energy electrons (LEEs) can initiate single- and double-strand breaks in plasmid DNA (deoxyribonucleic acid) even though their energies are well below the ionization threshold of DNA (about 7.5 eV). A key step in this LEE-induced DNA damage is thought to involve electron trapping on nucleobases2,4,5 which are the fundamental building blocks of DNA and RNA (ribonucleic acid). In this case, a lot of extensive studies6–13 have focused on the interactions between electrons and nucleobases. Furthermore, the electron affinity (EA) of a molecule is an important thermodynamic parameter reflecting its electron capturing ability. Thus, the researchers try to investigate the electron affinities of nucleobases or larger DNA and RNA subunits to understand the mechanism of radiation-induced DNA damage.
The adiabatic electron affinities (AEAs) of five DNA and RNA bases, namely, cytosine (C), uracil (U), adenine (A), guanine (G) and thymine (T), have been examined by lots of groups.14–20 A degree of agreement has emerged, whereby the AEA values of five nucleobases are in the range of −0.5 eV to 0.5 eV and the order of them is thought to be roughly U ≈ T > C > A > G. Among the five nucleobases, cytosine, which occurs naturally in DNA or RNA, is the most alkaline in aqueous solution (pKa = 4.6). It always chemically binds to a sugar moiety and interacts with other nucleic acid bases via hydrogen bonds. The first empirical estimates for the AEA value of cytosine was derived by Chen et al.,17 using substitution and replacement rules. They proposed that the value is calculated to be 0.6 eV or larger. Soon afterwards, the similar results were obtained by the both scaled reversible reduction potentials18,19 and Austin model 1 (AM1) semiempirical method.18,19 In recent years, Bowen's group14 developed experimental methods for successfully bringing parent nucleobases into the gas phase as valence state anions, and they extracted EA values of cytosine from the photoelectron spectra. Cytosine(H2O)n (n = 1–5) and their radical anions have been studied theoretically by Schaefer's group.21 They calculated the vertical detachment energies (VDEs) and adiabatic electron affinities of hydrated cytosine and predicted their prospective water-binding sites.
Comparing with cytosine, the interactions between electron and cytidine (composed of a cytosine and a sugar) have received less attention.22 This is because the choice of conformation for cytidine depends on the information sought, and some explanation as to the rationale for the initial geometries should be warranted. Therefore, the cytidine presents a conformational challenge. Bowen's group23 has performed an experimental study on the electrophilic properties of cytidine by measuring the anion photoelectron spectra of the parent (intact) nucleoside anions. Along with this investigation, we have systematically investigated the structures, electrophilic properties and hydrogen bonds of the neutral and anionic monohydrated cytidine (cytidine)H2O (ref. 24) in the last year. However, one bound water molecule could not be used to describe the real microhydration effects sufficiently. In aqueous solution, the interaction between water and nucleobases may cause significant changes in the structures and properties of the solute molecules compared to those in the gas phase. In a photodetachment-photoelectron spectroscopy study, Schiedt et al.25 proposed that the electron affinities of cytosine which arise from valence anionic states increase linearly with the number of hydrating water molecules. Predictably, the structures and properties of cytidine may also change with the number of hydrating water molecules. In the present study, we have investigated the effect of microsolvation by explicitly considering various structures of cytidine hydrated with up to four water molecules. The main objective is to investigate the effect of hydrating water molecules on the electrophilic properties of cytidine, meanwhile, to predict the favorable water-binding sites and the effect of extra electron attachment. Additionally, the weak interactions (especially hydrogen bonding) and charge distributions are also studied.
2. Computational methods
Geometry optimizations and frequency analyses of neutral and anionic cytidine(H2O)n (n = 1–4) have been carried out in the framework of density functional theory (DFT) method using the GAUSSIAN 03 program.26 B3LYP density functional, which is Becke's three-parameter functional27 (B3) in conjunction with the correlation functional of Lee, Yang, and Parr (LYP),28 was employed in combination with the 6-311G(2d,2p)29,30 basis set for studying this system. It has been demonstrated that this level of theory is very reliable in predicting the acidities of nucleobases31–33 as well as the geometrical and vibrational features of their hydrated complexes.34–36 Furthermore, in our previous study, we have performed a series of tests to confirm the reliability of our chosen computational method and basis set.23 The results based on B3LYP/6-311G(2d,2p) level are in good agreement with the experimental values.23
To search for various structures of our complexes, we used the fully optimized geometries of the pure neutral and anionic cytidine (Fig. 1) as the starting point. We wrote a shell script that randomly places one water molecule around the bare neutral and anionic cytidine fixed at the center of a sphere, with a restriction that all atoms in the water molecule should reside inside the sphere and that no atoms in the water molecule have too close contact (≤1 Å) with any atoms in the nucleoside. All the possible hydrogen bond forces and possible water-binding sites were considered. Furthermore, the previous studies21,37 on hydrated cytosine and thymidine were also employed as a guide. Then, we used the fully optimized stable structures of monohydrated cytidine as the starting point and placed another water molecule around them randomly, with the same restriction. Based on this method, cytidine trihydrates and tetrahydrates were also obtained gradually. In order to confirm that the optimized geometry corresponds to a local minimum in potential energy, each of them was followed by an analysis of harmonic vibrational frequencies. In this study, we predicted the vertical detachment energy (VDE), electron affinity (AEA and VEA) and hydration energies (Ehyd) for the studied complexes at B3LYP/6-311G(2d,2p) level. They are given by the following definitions:
| VDE = Eneutral at optimized anion geometry − Eoptimized anion |
| AEA = Eoptimized neutral − Eoptimized anion |
| VEA = Eoptimized neutral − Eanion at optimized neutral geometry |
| Ehyd = Ecytidine + nEwater − Ehydrated cytidine |
 |
| | Fig. 1 Molecular structures of cytidine with bond lengths in angstrom (black for the optimized neutral structure and red italic for the optimized anion structure). | |
The calculated results and some experimental values are listed in Table 1 and 2.
Table 1 Relative energies (ΔE), vertical electron affinities (VEAs), hydration energies (Ehyd) and intermolecular contacts for the neutral hydrated cytidine(H2O)n (n = 2–4). The numbering of the heteroatoms of cytidine and water is presented in Fig. 1
| Structure |
ΔE (eV) |
VEA (eV) |
Ehyd (kcal mol−1) |
Intermolecular contacts |
| Cw2-Na |
0.00 |
−0.39 |
27.54 |
N3⋯Hw–Ow |
| Ow⋯H–N4 |
| Cw2-Nb |
0.17 |
−0.72 |
23.60 |
Ow⋯H–O5′ |
| O5′⋯Hw–Ow |
| Cw2-Nc |
0.21 |
−0.49 |
22.74 |
N3⋯Hw–Ow⋯H–N4 |
| Ow⋯H–O5′ |
| Cw2-Nd |
0.22 |
−0.50 |
22.46 |
N3⋯Hw–Ow⋯H–N4 |
| O4′⋯Hw–Ow⋯H–O5′ |
| Cw2-Ne |
0.23 |
−0.46 |
22.13 |
N3⋯Hw–Ow⋯H–N4 |
| O3′⋯Hw–Ow |
| Cw3-Na |
0.00 |
−0.22 |
40.56 |
N3⋯Hw–Ow |
| Ow⋯H–N4, O2⋯Hw–Ow |
| Cw3-Nb |
0.01 |
−0.34 |
40.45 |
N3⋯Hw–Ow |
| Ow⋯H–N4 |
| Cw3-Nc |
0.15 |
−0.45 |
37.08 |
N3⋯Hw–Ow |
| Ow⋯H–N4, Ow⋯H–O5′ |
| Cw3-Nd |
0.16 |
−0.39 |
37.02 |
N3⋯Hw–Ow |
| Ow⋯H–N4, Ow⋯H–N4 |
| Cw3-Ne |
0.16 |
−0.62 |
36.90 |
N3⋯Hw–Ow⋯H–N4 |
| Ow⋯H–O5′, O5′⋯Hw–Ow |
| Cw4-Na |
0.00 |
−0.29 |
53.45 |
N3⋯Hw–Ow |
| Ow⋯H–N4, O2⋯Hw–Ow |
| Cw4-Nb |
0.01 |
−0.18 |
53.35 |
N3⋯Hw–Ow |
| Ow⋯H–N4 |
| Cw4-Nc |
0.04 |
−0.54 |
52.49 |
N3⋯Hw–Ow⋯H–N4 |
| Ow⋯H–O5′, O5′⋯Hw–Ow |
| Cw4-Nd |
0.10 |
−0.21 |
51.11 |
N3⋯Hw–Ow |
| Ow⋯H–N4, O2⋯Hw–Ow |
| Cw4-Ne |
0.11 |
|
50.95 |
O2′⋯Hw–Ow⋯H–O3′ |
| N3⋯Hw–Ow, O2′⋯H–Ow |
| Ow⋯H–N4, O2⋯Hw–Ow |
Table 2 Relative energies (ΔE), vertical electron affinities (VEAs), hydration energies (Ehyd) and intermolecular contacts for the anionic hydrated cytidine(H2O)n (n = 2–4). The numbering of the heteroatoms of cytidine and water is presented in Fig. 1
| Structure |
ΔE (eV) |
VDE (eV) |
Ehyd (kcal mol−1) |
Intermolecular contacts |
| Calc. |
Exp.23 |
| Cw2-Aa |
0.00 |
1.62 |
1.7 |
33.79 |
N3⋯Hw–Ow |
| O2⋯Hw–Ow |
| Cw2-Ab |
0.02 |
1.61 |
33.28 |
N3⋯Hw–Ow |
| O2⋯Hw–Ow |
| Cw2-Ac |
0.07 |
1.60 |
32.07 |
O2⋯Hw–Ow–Hw⋯N3 |
| O2′⋯H–Ow |
| Cw2-Ad |
0.12 |
1.54 |
31.05 |
N3⋯Hw–Ow⋯H–N4 |
| Ow⋯H–O5′ |
| Cw2-Ae |
0.17 |
1.53 |
29.92 |
N3⋯Hw–Ow⋯H–N4 |
| O2⋯Hw–Ow |
| Cw3-Aa |
0.00 |
1.63 |
— |
49.98 |
N3⋯Hw–Ow |
| Ow⋯H–N4, O2⋯Hw–Ow |
| Cw3-Ab |
0.11 |
1.55 |
47.40 |
N3⋯Hw–Ow |
| Ow⋯H–N4 |
| Cw3-Ac |
0.15 |
1.65 |
46.61 |
N3⋯Hw–Ow |
| Ow⋯H–N4, Ow⋯H–O5′ |
| Cw3-Ad |
0.18 |
1.64 |
45.75 |
N3⋯Hw–Ow |
| Ow⋯H–N4, Ow⋯H–N4 |
| Cw3-Ae |
0.21 |
1.79 |
45.16 |
N3⋯Hw–Ow⋯H–N4 |
| O2⋯Hw–Ow, O2⋯Hw–Ow |
| Cw4-Aa |
0.00 |
1.77 |
— |
66.58 |
N3⋯Hw–Ow |
| Ow–H⋯N4 |
| Cw4-Ab |
0.10 |
1.71 |
64.34 |
N3⋯Hw–Ow |
| Ow⋯H–N4, O2⋯Hw–Ow |
| Cw4-Ac |
0.15 |
1.88 |
62.97 |
N3⋯Hw–Ow |
| Ow–H⋯N4, O2⋯Hw–Ow |
| Cw4-Ad |
0.20 |
1.72 |
61.79 |
N3⋯Hw–Ow |
| Ow⋯H–N4, O2⋯Hw–Ow |
| O2′⋯Hw–Ow⋯H–O3′ |
| Cw4-Ae |
0.21 |
1.73 |
61.78 |
N3⋯Hw–Ow, O2′⋯H–Ow |
| Ow⋯H–N4, O2⋯Hw–Ow |
3. Results and discussion
3.1. Geometrical structures
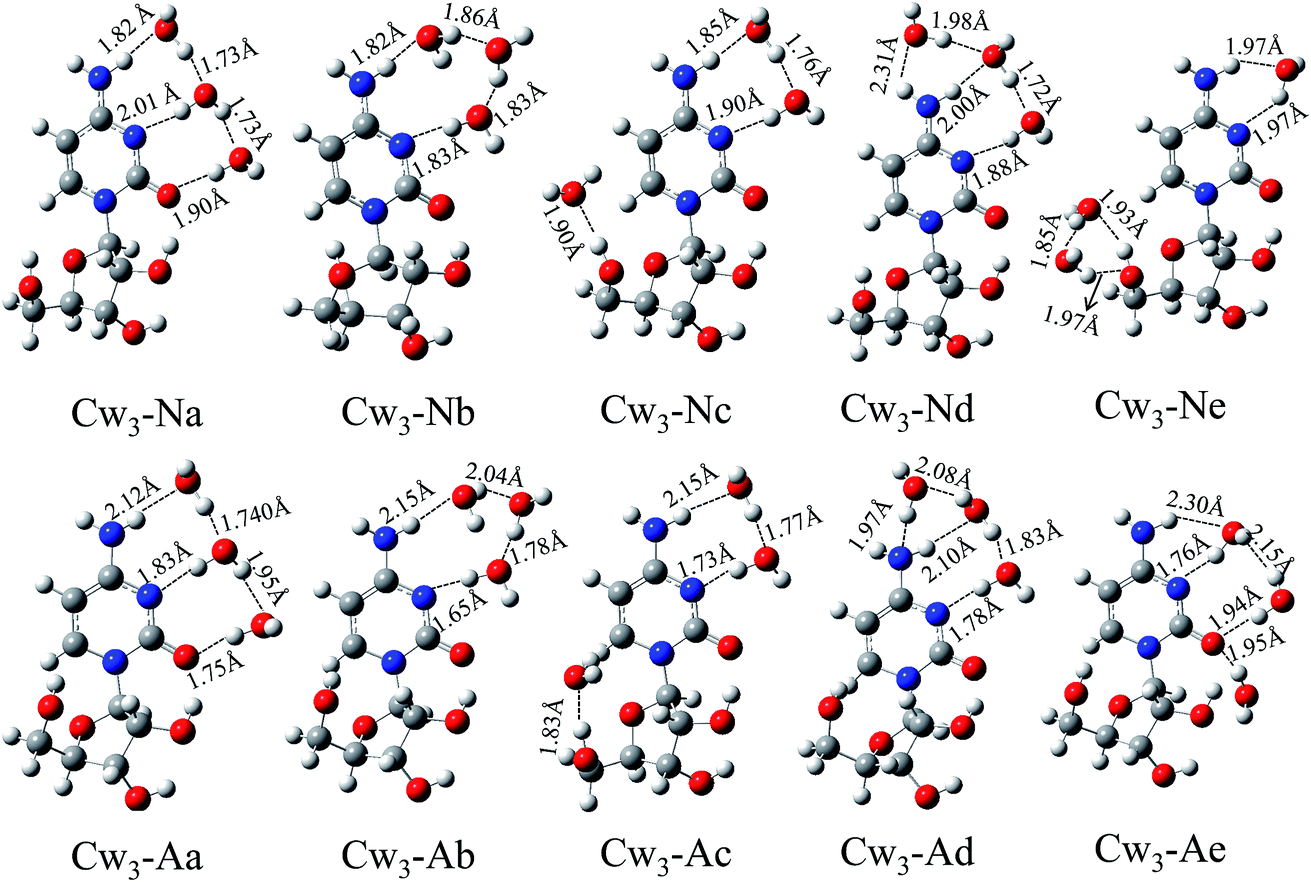
Based on the method mentioned above, a large number of optimized isomers for hydrated cytidine have been obtained. We only select five energetically low-lying isomers for each type complexes and list them together with the hydrogen bond length in Fig. 2–4, respectively. In addition, the next five low-lying isomers are listed in ESI (Fig. S2–S4†). For convenience, the neutral and anionic hydrated cytidines are designated by CWn-N and CWn-A, respectively, where n is the number of hydrating water molecules. They are followed by a lowercase letter representing their energies from low to high. The numbering of the heteroatoms of cytidine and water is presented in Fig. 1. Their corresponding relative energies (ΔE), vertical detachment energies (VDEs), vertical electron affinities (VEAs), hydration energies (Ehyd) and intermolecular contacts between nucleoside and water are summarized in Table 1 and 2. In addition, the low-lying structures and the corresponding information of cytidine monohydrates are listed in ESI.†
 |
| | Fig. 2 Molecular structures for cytidine dihydrates (first line) and their anions (second line), with intermolecular distances in angstrom. The blue, red, grey and white balls represent N, O, C and H atoms, respectively. | |
 |
| | Fig. 3 Molecular structures for cytidine trihydrates (first line) and their anions (second line), with intermolecular distances in angstrom. The blue, red, grey and white balls represent N, O, C and H atoms, respectively. | |
 |
| | Fig. 4 Molecular structures for cytidine tetrahydrates (first line) and their anions (second line), with intermolecular distances in angstrom. The blue, red, grey and white balls represent N, O, C and H atoms, respectively. | |
3.1.1. Cytidine dihydrates and their anions. Five low-lying energy structures for the neutral cytidine dihydrates, along with their respective anions, are displayed in Fig. 2. The lowest-energy structure CW2-Na has the same water-binding site as the lowest-energy neutral cytidine monohydrates.24 In structure CW2-Na, two water molecules bind to cytosine base unit of cytidine via the N3⋯Hw–Ow and Ow⋯H–N4 hydrogen bonds. Meanwhile, a hydrogen bond is also formed between the two water molecules resulting in a long cyclic bond. The binding energy of this cyclic bond formation is calculated to be −27.54 kcal mol−1. Structures CW2-Nb has both the two water molecules binding to the sugar moiety. CW2-Nc is a very interesting structure, in which both the cytosine base and sugar unit are associated with water molecule via the N3⋯Hw–Ow⋯H–N4 and Ow⋯H–O5′ hydrogen bond. CW2-Nd has a similar structure as CW2-Nc. The difference between them is that one water molecule bind to sugar unit via Ow⋯H–O5′ single hydrogen bond in C2W-Nc, while it is a cyclic hydrogen bond O4′⋯Hw–Ow⋯H–O5′ in CW2-Nd. Likely, two water molecules bind to both the cytosine base and sugar unit in structure Cw2-Ne, while the sugar unit is associated with water molecule via the O3′⋯Hw–Ow bond. In addition, the VEA values for these five dihydrated cytidine structures range from −0.39 to −0.72 eV.As for cytidine(H2O)2−, most of the low-lying structures are very different from those of the neutrals. Only Cw2-Ad has the similar water-binding site with Cw2-Nd structure. This indicates that the attachment of the extra electron has significant effect on the hydrogen bonds. The Ow–Hw⋯N3 hydrogen bond of CW2-Ad is shorter by 0.167 Å than that of its corresponding neutral (CW2-Nd), while the Ow⋯H–N4 hydrogen bond is longer by 0.194 Å. The explanation could be that the excess negative charge density is mainly localized on the cytidine moiety. If a nitrogen or oxygen atom of cytosine acts as hydrogen bond acceptor, there is an increase in the negative charges of H-bond acceptor upon the attachment of extra electron.38 These may make the electron density easier to transfer from the cytosine moiety to the hydrogen atom of water molecule. The resulting hydrogen bond in a cytidine monohydrate anion would be strengthened and shortened compared to that of the corresponding neutral. On the other hand, a hydrogen bond where the oxygen atom of the water molecule acts as a hydrogen bond acceptor would be weakened because the transfer of negative charge density from water to cytidine is unfavorable. In structure CW2-Aa, two water molecules and cytidine bind together via three hydrogen bonds: N3⋯Hw–Ow, O2⋯Hw–Ow and Ow⋯Hw–Ow. The water-binding sites of CW2-Ab are similar with those of Cw2-Aa, and even the hydrogen bond lengths are almost equal. The only difference between them is in the water–water hydrogen bond, in which the role of hydrogen bonding donors and acceptor is interchange. In structures Cw2-Ac and Cw2-Ad, the two water molecules bind to both the cytosine base and sugar unit. The computed VDEs for these dihydrated structures are in the range of 1.53 to 1.62 eV, which is consistent with the experimental value (1.7 eV) obtained by Bowen and co-workers.23
3.1.2. Cytidine trihydrates and their anions. Based on the cytidine monohydrates and dihydrates, lots of structures for cytidine trihydrates have been obtained. We only select five energetically low-lying isomers and list in Fig. 3. As can be seen from Fig. 3, the lowest energy structures Cw3-Na can be obtained by adding one water molecule to Cw2-Na. Comparing the cytidine(H2O)0/−, cytidine(H2O)20/− and cytidine(H2O)30/− systematically, we find that the most favorable water-binding sites of cytidine are N3⋯H–N4 and O2. The water molecules of Cw3-Nb, Cw3-Nc, Cw3-Nd and Cw3-Ne all occupy these three sites. Structures Cw3-Na and Cw3-Nb are almost degenerate in total energy. Accordingly, their hydration energies are almost equal (40.56 kcal mol−1 and 40.45 kcal mol−1). In structures Cw3-Nc and Cw3-Ne, the water molecules bind to both the cytosine base and sugar unit, but the number of water molecules binding to two units is different. There are both cyclic and single hydrogen bonds in isomer Cw3-Nd.Unlike the cytidine dihydrates, most of the neutrals and anions of cytidine trihydrates have similar structures, such as Cw3-Na and Cw3-Aa, Cw3-Nb and Cw3-Ab, Cw3-Nc and Cw3-Ac, Cw3-Nd and Cw3-Ad. However, the hydrogen bond lengths of neutral and anionic complexes are different. Upon the extra electron attachment, the hydrogen bond where the hydrogen atom of the water molecule acts as a hydrogen bond donor would be shortened, while the hydrogen bond where the oxygen atom of the water molecule acts as a hydrogen bond acceptor would be elongated. Structure Cw3-Ae involves three hydrogen bonds: N3⋯Hw–Ow⋯H–N4, Ow⋯H–O5′ and O5′⋯Hw–Ow. Furthermore, the calculated VDEs of these five isomers are in the range from 1.63 eV to 1.79 eV. Unfortunately, there is no available experimental value until now. We hope that our studies might offer information for further investigations and facilitate the experiment in future.
3.1.3. Cytidine tetrahydrates and their anions. Fig. 4 shows the low-lying structures of tetrahydrated cytidine and their anions. As can be seen from the figures, most of the structures can be obtained by adding one or two water molecules on cytidine(H2O)20/− or cytidine(H2O)30/− isomers. For example, in the lowest energy neutral tetrahydrates Cw4-Na, the fourth water molecule is associated with Cw3-Nb through water–water hydrogen bond. It is noteworthy that the fourth water molecule binds to the previous water molecule in the first three low-lying structures of both neutral and anionic cytidine(H2O)4. Only in Cw4-Nd, Cw4-Ne, Cw4-Ad and Cw4-Ae, the four water molecules form a compact hydrogen bond network around the cytidine molecule. Throughout all the structures of both neutral and anionic cytidine(H2O)4, most of the water molecules tend to bind with the cytosine base. Structures Cw4-Nb, Cw4-Nd and Cw4-Ne have the same water-binding sites with Cw4-Ab, Cw4-Ad and Cw4-Ae, respectively.
3.1.4. Adiabatic electron affinities. In our previous study,24 the adiabatic electron affinities (AEAs) of cytidine and cytidine(H2O) are calculated to be 0.17 and 0.34 at B3LYP/6-311G(2d,2p) level. In the present study, the AEAs of cytidine(H2O)2, cytidine(H2O)3 and cytidine(H2O)4 are calculated to be 0.44 eV, 0.58 eV and 0.68 eV, respectively. The AEA values of cytidine(H2O)n against the corresponding number of water molecules are plotted in Fig. 5. From the figure, we can clearly see that their AEAs increase linearly with the number of hydrating water molecules. This indicates that the cytidine(H2O)n would get a stronger ability to attract electron with the hydration number increasing. On the other hand, Schiedt et al.25 proposed that the electron affinities of cytosine which arise from valence anionic states increase linearly with the number of hydrating water molecules. The electron affinities for both cytidine and cytosine have the similar evolution tendencies against the hydration number.
 |
| | Fig. 5 Adiabatic electron affinities (AEAs) for cytidine hydrates with up to four water molecules. | |
3.2. Charge distributions
Upon extra electron attachment, the “surplus electron” qualitatively occupies the lowest unoccupied molecular orbital (LUMO) of neutral structure. Thus the singly occupied molecular orbital (SOMO) of the corresponding anion can reveal the character of the “surplus electron” distribution. In the present study, we have examined the characteristics of the SOMOs for hydrated cytidine anions. The plots of SOMOs are listed in Fig. 6. As can be seen from Fig. 6, there is a consensus across all the SOMOs. Almost all of the orbital density localized on cytidine, especially on cytosine base. This indicates that the excess electron density is localized on the cytidine moiety, especially on the cytosine base unit. The water molecules have no excess negative charge density localized because of its negative AEAs. The results are consistent with our previous study on monohydrated cytidine.
 |
| | Fig. 6 Singly occupied molecular orbitals (SOMOs) for the anions of cytidine(H2O)n (n = 2–4). The isovalue is 0.02 a.u. | |
In order to probe into the distribution of electron density and the reliable charge-transfer information induced by the extra electron attachment, we have calculated the natural population analysis (NPA) for neutral and anionic cytidine(H2O)n (n = 2–4) complexes. While we only show the atomic charges of the first two lowest energy structures of each type cytidine hydrates in Fig. S6–S8.† In the lowest-energy structure of cytidine dihydrates (Cw2-Na), both of the two water molecules possesses 0.01e positive charges. This indicates that a little of electron density transfers from water molecules to cytidine moiety. The atomic charges of oxygen and two hydrogen atoms in these two water molecules are calculated to be (−0.95e, 0.46e, 0.49e) and (−0.95e, 0.46e, 0.50e), respectively. Comparing with a free water molecule (−0.89e, 0.44e and 0.44e), we can see that the charges of water oxygen and hydrogen atoms, which form hydrogen bonds with cytidine, show an obvious change. Due to the structures Cw3-Na and Cw3-Aa have the same water-binding sites, we take them as example to elucidate the distribution of excess negative charge. In isomer Cw3-Na, the total charges of three water molecules are virtually zero. Upon an extra electron attach to structure Cw3-Na, their charges change to be −0.03e, −0.03e and −0.01e respectively in anion Cw3-Aa. From this we can see that the total excess negative charge localized on the three water molecules is only −0.07e. That is, the extra electron is mainly localized on the cytidine moiety, especially on cytosine. This is in agreement with the result obtained by analyzing SOMOs. In addition, when the extra electron attach to cytidine, the charges on N3 nitrogen atom of Cw3-Aa increase from −0.64e to −0.71e, and that of the O2 nitrogen atom increase from −0.76e to −0.83e. On the other hand, the charges on N4–H hydrogen atom decrease from 0.44e to 0.39e. These may make the electron density easier to transfer from the cytidine moiety to the hydrogen atom of water molecule. The transfer of negative charge density from the water oxygen atom, which acts as hydrogen bond acceptor, to the N4–H hydrogen atom is more difficult. Thus, the attachment of the extra electron shortens the Ow–Hw⋯N3 and Ow–Hw⋯O2 hydrogen bonds and elongates the Ow⋯H–N4 hydrogen bond. This verifies the explanation of the structural changes as revealed in the discussion of the geometrical structures.
3.3. Weak interaction in hydrated cytidine
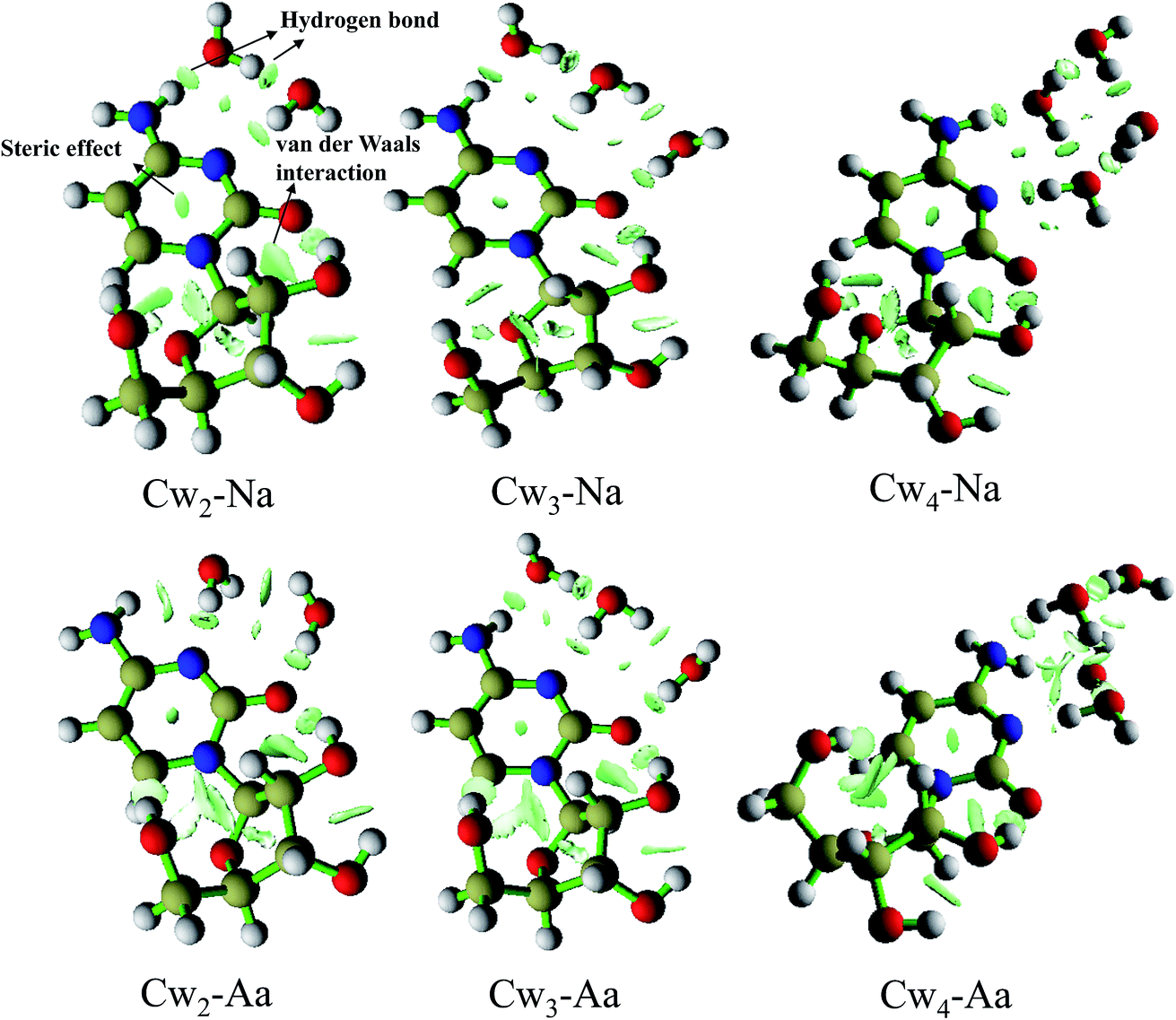
Weak interactions, including hydrogen bond, electrostatic interactions, van der Waals interactions, steric repulsion and so on, are the driving force in most biochemical processes. In particular, the hydrogen bonds are fundamental in determining biomolecular structure and play important role in DNA replication, gene expression, and DNA repair. Thus the ability to understand and predict the formation of weak interactions is of great relevance to a diversity of problems in science. In this section, we present an approach to map and analyze the weak interaction of hydrated cytidine, especially hydrogen bond. This approach depends on the electron density and reduced density gradient (RDG), and it has been successfully applied to weak interaction by Yang group.39,40 RDG is a fundamental dimensionless quantity in DFT used to describe the deviation from a homogeneous electron distribution.41,42
To show a rich visualization of weak interactions in cytidine(H2O)n0/− (n = 2–4), we plot the isosurfaces of RDG for the lowest energy structures in Fig. 7. Isosurfaces are generated for 0.5 a.u. The unwanted chemical bonds and around nuclei regions have been shielded and only the isosurfaces of weak interactions are eventually remained. Here, we take the structure Cw2-Na as an example to expatiate the characteristic of those plots. As shown in Fig. 7, the different types of weak interactions have been clearly shown and identified in this map. Obviously, the elliptical slab between oxygen (nitrogen) and hydrogen atoms is hydrogen bond. Both of the intermolecular and intramolecular hydrogen bonds can be clearly seen in this plot. The interaction between O2 and C2′ can be identified as van der Waals interaction. In the center of rings, the strong steric effect (also called nonbond overlap) has been marked. It worth pointing out that the steric effect originates from the fact that each atom in a molecule occupies a certain amount of space. It is closely associated with the quantum effects, which come from the Pauli exclusion principle and dynamic electron correlation effect, preventing both same-spin and opposite-spin electrons from coming together. As for the plots of cytidine trihydrates and tetrahydrates, they have the similar characteristic of weak interactions shape as that of Cw2-Na.
 |
| | Fig. 7 Reduced density gradient isosurfaces (0.5 a.u.) for the lowest-energy structures of cytidine hydrates and their anions. The blue, red, yellow and white balls represent N, O, C and H atoms, respectively. | |
4. Conclusions
Microhydration effects on the geometrical structures, electron affinities and charge distributions of cytidine and its anions have been investigated systematically. All the results are summarized as follows.
(1) Based on geometry optimization, various structures of cytidine(H2O)n0/− (n = 2–4) have been predicted. By comparing the lowest energy structures of each type hydrates, N3, H–N4 and O2 are found to be the most favorable water-binding sites of cytidine. Upon the extra electron attachment, the hydrogen bond where the hydrogen atom of the water molecule acts as a hydrogen bond donor would be elongated, while the hydrogen bond where the oxygen atom of the water molecule acts as a hydrogen bond acceptor would be shortened.
(2) The predictions of electron affinities and vertical detachment energies are carried out to investigate the electrophilic properties of cytidine hydrates. The AEAs of cytidine(H2O)n increase linearly with the increasing number of hydrating water molecules, indicating that they would get a stronger ability to attract electron with the hydration number increasing.
(3) By examining the SOMO and natural population analysis, we found the excess electron density is localized on the cytidine moiety, especially on the cytosine base unit. This may help explain why the hydrogen bond changes upon the extra electron attachment. In addition, we present an approach to map and analyze the weak interaction based on the electron density (ρ) and reduced density gradient (RDG). From RDG isosurfaces, we can see the hydrogen bond is formed between oxygen (nitrogen) and hydrogen atoms. The steric effect can be clearly identified in the center of rings.
Acknowledgements
This work was supported by the Special Foundation for Theoretical Physics Research Program of China (No. 11547147), Natural Science Foundations of Shaanxi Province (No. 2016JQ1003 and 2016JQ1028), Scientific Research Plan Projects of Shaanxi Education Department (No. 16JK1098) and the Shaanxi University of Science & Technology Key Research Grant (No. BJ15-07 and 2016BJ-01).
References
- B. Boudaïffa, P. Cloutier, D. Hunting, M. A. Huels and L. Sanche, Science, 2000, 287, 1658–1660 CrossRef.
- L. Sanche, Mass Spectrom. Rev., 2002, 21, 349–369 CrossRef CAS PubMed.
- F. Martin, P. D. Burrow, Z. Cai, P. Cloutier, D. Hunting and L. Sanche, Phys. Rev. Lett., 2004, 93, 068101 CrossRef PubMed.
- H. Abdoul-Carime, S. Gohlke and E. Illenberger, Phys. Rev. Lett., 2004, 92, 168103 CrossRef PubMed.
- I. Anusiewicz, J. Berdys, M. Sobczyk, P. Skurski and J. Simons, J. Phys. Chem. A, 2004, 108, 11381–11387 CrossRef CAS.
- G. Hanel, S. Denifl, P. Scheier, M. Probst, B. Farizon, M. Farizon, E. Illenberger and T. D. Märk, Phys. Rev. Lett., 2003, 90, 188104 CrossRef CAS PubMed.
- C. Desfrançois, H. Abdoul-Carime and J. P. Schermann, J. Chem. Phys., 1996, 104, 7792 CrossRef.
- M. Haranczyk and M. Gutowski, J. Am. Chem. Soc., 2005, 127, 699–705 CrossRef CAS PubMed.
- R. A. Bachorz, J. Rak and M. Gutowski, Phys. Chem. Chem. Phys., 2005, 7, 2116–2125 RSC.
- X. Bao, H. Sun, N. B. Wong and J. Gu, J. Phys. Chem. B, 2006, 110, 5865–5874 CrossRef CAS PubMed.
- M. Haranczyk, M. Gutowski, X. Li and K. H. Bowen, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 4804–4807 CrossRef CAS PubMed.
- J. Simons, Acc. Chem. Res., 2006, 39, 772–779 CrossRef CAS PubMed.
- M. Haranczyk, M. Gutowski, X. Li and K. H. Bowen, J. Phys. Chem. B, 2007, 111, 14073–14076 CrossRef CAS PubMed.
- X. Li, K. H. Bowen, M. Haranczyk, R. A. Bachorz, K. Mazurkiewicz, J. Rak and M. Gutowski, J. Chem. Phys., 2007, 127, 174309 CrossRef PubMed.
- S. Eustis, D. Wang, S. Lyapustina and K. H. Bowen, J. Chem. Phys., 2007, 127, 224309 CrossRef PubMed.
- R. A. Bachorz, W. Klopper, M. Gutowski, X. Li and K. H. Bowen, J. Chem. Phys., 2008, 129, 054309 CrossRef PubMed.
- E. C. M. Chen, E. S. D. Chen and W. E. Wentworth, Biochem. Biophys. Res. Commun., 1990, 171, 97–101 CrossRef CAS PubMed.
- J. R. Wiley, J. M. Robinson, S. Ehdaie, E. C. M. Chen, E. S. D. Chen and W. E. Wentworth, Biochem. Biophys. Res. Commun., 1991, 180, 841–845 CrossRef CAS PubMed.
- E. S. D. Chen, E. C. M. Chen, N. Sane and S. Shulze, Bioelectrochem. Bioenerg., 1999, 48, 69–78 CrossRef CAS PubMed.
- Q. Zhang and E. C. M. Chen, Biochem. Biophys. Res. Commun., 1995, 217, 755–760 CrossRef CAS PubMed.
- S. Kim and H. F. Schaefer III, J. Chem. Phys., 2007, 126, 064301 CrossRef PubMed.
- C. A. Schroeder, E. Pluhařová, R. Seidel, W. P. Schroeder, M. Faubel, P. Slavíček, B. Winter, P. Jungwirth and S. E. Bradforth, J. Am. Chem. Soc., 2014, 137, 201–209 CrossRef PubMed.
- X. Li, H. Wang and K. H. Bowen, J. Chem. Phys., 2010, 133, 144304 CrossRef PubMed.
- P. Shao, X. Y. Kuang, L. P. Ding and Y. R. Zhao, J. Chem. Phys., 2013, 139, 024305 CrossRef PubMed.
- J. Schiedt, R. Weinkauf, D. M. Neumark and E. W. Schlag, Chem. Phys., 1998, 239, 511–524 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery Jr, T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. G. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 09, Revision C.0, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650 CrossRef CAS.
- W. J. Hehre, L. Radom. P. V. R. Schleyer and J. A. Pople, Ab initio molecular Orbital Theory, Wiley, New York, 1986 Search PubMed.
- M. A. Kurinovich and J. K. Lee, J. Am. Chem. Soc., 2000, 122, 6258–6262 CrossRef CAS.
- A. Hocquet and M. Ghomi, Phys. Chem. Chem. Phys., 2000, 2, 5351–5353 RSC.
- Y. Huang and H. Kenttamaa, J. Phys. Chem. A, 2004, 108, 4485–4490 CrossRef CAS.
- A. K. Chandra, M. T. Nguyen and T. Zeegers-Huyskens, J. Chem. Soc., Faraday Trans., 1998, 94, 1277–1280 RSC.
- A. K. Chandra, M. T. Nguyen and T. Zeegers-Huyskens, J. Phys. Chem. A, 1998, 102, 6010–6016 CrossRef CAS.
- K. C. Hunter, L. R. Rutledge and S. D. Wetmore, J. Phys. Chem. A, 2005, 109, 9554–9562 CrossRef CAS PubMed.
- S. Kim and H. F. Schaefer III, J. Chem. Phys., 2010, 133, 144305 CrossRef PubMed.
- M. Dreyfus and A. Pullman, Theor. Chim. Acta, 1970, 19, 20–37 CrossRef CAS.
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed.
- J. Contreras-García, W. Yang and E. R. Johnson, J. Phys. Chem. A, 2011, 115, 12983–12990 CrossRef PubMed.
- P. Hohenberg and W. Kohn, Phys. Rev. [Sect.] B, 1964, 136, 864–871 CrossRef.
- A. J. Cohen, P. Mori-Sánchez and W. Yang, Science, 2008, 321, 792–794 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra11720a |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.