
A facile construction of Au nanoparticles on a copolymer ligand brushes modified graphene oxide nanoplatform with excellent catalytic properties†
Yajiao Songa,
Jianhua Lüa,
Bingxin Liub and
Changli Lü*a
aKey Laboratory of Nanobiosensing and Nanobioanalysis at Universities of Jilin Province, Department of Chemistry, Northeast Normal University, Changchun 130024, P. R. China. E-mail: lucl055@nenu.edu.cn
bSchool of Mechanical Engineering, Qinghai University, Xining 810016, P. R. China
First published on 4th July 2016
Abstract
In this paper, we described an effective method for the in situ synthesis of Au nanoparticles (Au NPs) on 8-hydroxyquinoline ligands-containing copolymer brushes modified graphene oxide (GO) nanosheets. The copolymer brushes of P(OEGMA-co-MQ) were grafted from the surface of GO by reversible addition–fragmentation chain transfer (RAFT) polymerization based on the monomers of oligo(ethylene glycol) monomethyl ether methacrylate (OEGMA) and 5-(2-methacryloyl-ethyloxymethyl)-8-quinolinol (MQ). Au NPs were generated via in situ reduction of Au3+ by an alkali and the 8-hydroxyquinoline units in the copolymer brushes were used as capping agents to stabilize Au NPs. The obtained Au NPs–GO hybrids exhibited a weak blue fluorescence emission at 475 nm and high catalytic activity in water for the reduction of 4-nitrophenol (4-NP).
1. Introduction
Graphene, as a newly discovered two-dimensional nanomaterial composed of sp2-bonded carbon atoms, has attracted intense interest since monolayer graphene was successfully fabricated in 2004.1,2 Due to its fascinating optical, electronic, thermal, and mechanical properties which arise from its honeycomb structure, graphene has been incorporated into many composite materials to explore their applications in various fields of science and engineering.3 Meanwhile, graphene oxide (GO), a derivative of graphene, has also been widely researched in recent years because of its convenient synthetic method on a large-scale and abundance of oxygen-containing functional groups on the surface.1,4 The abundant oxygen-containing functional groups such as epoxide, alcohol and carboxylic acids not only provide GO with excellent aqueous dispersity, but also convey a remarkable opportunity for further chemical modification, especially for chemically functionalization with polymers.4In the past several decades, the polymer functionalized GO for the fabrication of nanocomposites has drawn a great deal of attention. In general, the technique of polymer functionalized GO includes both covalent and non-covalent strategy. In the covalent modification, taking advantage of the rich chemistry of oxygen-containing functional groups to chemical attachment of small organic molecules on graphene oxide sheets has been exploited by the great majority.5 It brings sufficient opportunities for further modification with polymer by “grafting to” or “grafting from” techniques. The simple route of “grafting to” is the direct covalent linkage of the functional polymers on the GO surface using esterification,6 amidation,7 click chemistry,8 nitrene chemistry,9,10 radical addition11 and so on.12,13 The “grafting from” technique is associated with the polymerization of monomers from the macroinitiators derived from the surface of GO. The atom transfer radical polymerization (ATRP) and reversible addition–fragmentation chain transfer (RAFT) polymerization are two important methods employed for “grafting from” techniques.14,15 When a comparison is made between “grafting to” and “grafting from” techniques, it is found that the technique of “grafting from” is a better strategy for GO modification than “grafting to”. Because the “grafting from” route could provide high graft density and processability for the thin graphene oxide sheets in a quicker way.5 Apart from the covalent functionalization, the non-covalent modification on graphene surface has also been used significantly. For example, H-bonding16 and π–π stacking17 on the surface of GO play an important role in the non-covalent interactions.
Noble metal nanoparticles have attracted significant increasing attention over the past few years owing to their unique physical and chemical properties which are totally different from those of bulk metals.18 These properties make them become suitable materials for potential applications in various fields such as photonics,19 catalysis,4,20,21 chemical and biological sensing,22 information storage23 and surface enhance Raman scattering (SERS).1 Among these noble metal nanoparticles, gold nanoparticles (Au NPs) are widely reported because of their specific optical, electric, delivery and especially catalytic properties.20 Unfortunately, smaller Au NPs easily aggregate to minimize their surface area due to their higher surface energy, resulting in an obvious decrease in their catalytic activity.18 To address the disadvantage, numerous methods have been utilized.24,25 In these strategies, introducing metal nanoparticles on/into less expensive solid supports to form composite catalysts is regarded as an effective strategy to solve the problem.21 Graphene and his derivative of graphene oxide have been used as an outstanding support due to their unique structure and huge specific surface area.21 In recent years, plenty of reports have been devoted to the fabrication of Au NPs/graphene composite,3,4,20 but few reports of water-soluble GO were used as supports.1,21 On the other hand, although these graphene or graphene oxide based Au NPs nanocatalysts have excellent catalytic performance,4,18,21 it is an important and challenging work to explore the stable Au NPs/GO hybrid nanomaterials via coordinate bonds with functional ligands decorated graphene oxide.
Herein, we developed a novel strategy to construct Au NPs–GO hybrids nanomaterials based on 8-hydroxyquinoline ligand-containing copolymer brushes functionalized GO. Scheme 1 shows our key strategy for the fabrication of the copolymer brushes–GO hybrid via RAFT polymerization. Firstly, 2-[(dodecylsulfanyl)carbonothioyl sulfanyl]propanoic acid (RAFT agent) was functionalized on the surface of GO by esterification, and then the water-soluble copolymer of P(OEGMA-co-MQ) was grafted from the surface of GO sheets via RAFT polymerization. Then, Au NPs were generated on the copolymer brushes modified GO via in situ reduction of Au3+ by alkali and the 8-hydroxyquinoline units in the polymer brushes as capping agent to stabilize Au NPs. The optical and catalytic properties of the resulting Au NPs–GO hybrids were studied. It was found that the nanohybrids exhibited high catalytic activity in water for the reduction of 4-NP (Scheme 2).
 | ||
| Scheme 1 Schematic illustration for preparation of copolymer brushes–GO. | ||
 | ||
| Scheme 2 Schematic illustration for preparation of Au NPs–GO hybrid and its catalytic reduction of 4-NP. | ||
2. Experimental
2.1 Materials
Gold(III) chloride trihydrate (HAuCl4·3H2O, 99%) was obtained from Macklin. Graphite powder was purchased from Sigma-Aldrich. Oligo(ethylene glycol) monomethyl ether methacrylate (OEGMA) was obtained from Sigma-Aldrich and passed through an alumina column to remove the inhibitor before use. 2,2-Azoisobutyronitrile (AIBN) was recrystallized from ethanol (95%) before use. 2-[(Dodecylsulfanyl)carbonothioylsulfanyl]propanoic acid (RAFT agent)26 and 5-(2-methacryloylethyloxy-methyl)-8-quinolinol (MQ)27 were synthesized according to literature procedures. Graphene oxide was synthesized according to the previous report using the modified Hummers method.28 All other reagents were purchased from commercial sources and used as received.2.2 Preparation of copolymer brushes grafted GO (copolymer brushes–GO)
The RAFT agent was grafted from GO sheets via a N,N-dicyclohexylcarbodiimide (DCC) coupling reaction.15 Briefly, 50 mL of DMF solution containing GO (100 mg), RAFT agent (0.35 g) and 4-dimethylaminopyridine (DMAP, 0.06 g) was ultrasonicated for 30 min. After stirred for 1 h at 0 °C, DCC (0.2 g) was added into the mixture, followed by an additional reaction for 48 h at room temperature. The product (GO-RAFT) were centrifuged and washed with deionized water and THF respectively and then concentrated in vacuo.Typical procedure employed for the synthesis of P(OEGMA-co-MQ) grafted from GO was as follows. OEGMA (800 mg), MQ (46 mg), GO-RAFT (25 mg) and AIBN (2 mg) were charged into a three-necked round-bottom flask containing 3 mL dry DMF. The flask was degassed by three freeze–pump–thaw cycles and then sealed under vacuum. The polymerization was conducted at 70 °C in an oil bath. After 24 h, the mixture was centrifuged and washed with THF for several times and the copolymer brushes–GO hybrid was obtained after drying in vacuum overnight at room temperature. For comparison, the free copolymer P(OEGMA-co-MQ) was also prepared at the same condition, but the RAFT agent 2-[(dodecylsulfanyl)carbonothioylsulfanyl]propanoic acid was used to take the place of GO-RAFT. In order to determine the molecular weight of P(OEGMA-co-MQ) grafted from the surface of GO, 60 mg of P(OEGMA-co-MQ) grafted GO hybrid and 120 mg of m-chloroperbenzoic acid (MCPA) were mixed and stirred for 15 h at ambient temperature. After that, the mixture was centrifuged and the supernatant was collected. The P(OEGMA-co-MQ) copolymers removed from the surface of GO were precipitated into ether, and dried in vacuum at ambient temperature for 24 h. The molecular weight and polydispersity index of P(OEGMA-co-MQ) were determined by GPC using DMF as eluent (Mn = 62![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1, PDI = 1.35).
000 g mol−1, PDI = 1.35).
2.3 In situ synthesis of Au NPs on copolymer brushes modified GO (Au NPs–GO)
To generate Au NPs on the copolymer brushes–GO, different amount of HAuCl4·3H2O (0.5 and 0.7 mg) was added into 1 mL of copolymer brushes–GO aqueous solution (3 mg mL−1) respectively under vigorous stirring. After stirring for 1 h, the pH value of the mixture solution was adjusted to about 9 with ammonia solution (25%), and then the solution was stirred for another 2 h at room temperature. After that, the above mixture was centrifuged and washed with deionized water for several times. The Au NPs–GO hybrid-1 (0.5 mg HAuCl4·3H2O) and Au NPs–GO hybrid-2 (0.7 mg HAuCl4·3H2O) were obtained after drying under vacuum for 24 h. The Au contents in the above Au NPs–GO hybrid-1 and 2 are 6.5 and 8.9 wt% respectively. We also prepared Au NPs–GO hybrid-1 at other two pH values at 8 and 10 to verify their different fluorescent properties. In a control experiment, Au NPs were prepared by mixing HAuCl4·3H2O (0.5 mg) with the copolymer P(OEGMA-co-MQ) without the presence of GO at the same condition.2.4 Reduction of 4-nitrophenol catalyzed by Au NPs–GO hybrids
Typically, 1 mL solution of 4-nitrophenol (0.2 mM) and NaBH4 aqueous solution (0.3 mL, 20 mM) were incubated in the quartz cell (1 cm path length). After that, 30 μL of GO–Au NPs hybrids (3 mg mL−1) was added into the above mixture and immediately placed in the cell holder of the spectrophotometer. The catalytic reduction of 4-nitrophenol (4-NP) to 4-aminophenol (4-AP) by excess NaBH4 with the Au NPs–GO hybrids as a catalyst was monitored by the time-dependent adsorption spectra in a scanning range of 200–600 nm at ambient temperature.2.5 Measurements
Transmission electron microscopy (TEM) images were captured by JEOL-2021 electron microscope. UV-vis absorption spectra were collected in the range from 200 to 700 nm by using SHIMADZU UV-2550 UV-visible spectrophotometer. FT-IR spectra were performed by KBr disks on a Magna 560 FT-IR spectrometer. Photoluminescence (PL) properties were measured on a Cary Eclipse fluorescence spectrometer. The fluorescence emission spectra were measured in 400–700 nm with an excitation at 360 nm. Thermal gravimetric analysis (TGA) was carried out using a Perkin-Elmer Pyris 6 TGA instrument with a heating rate of 10 °C min−1 under nitrogen. The thermograms were obtained from ambient temperature to 700 °C and nitrogen was used as the purging gas. X-ray diffraction (XRD) spectra were collected on an X-ray diffraction instrument (Siemens D5000) with a Cu target (λ = 0.1540 nm) at room temperature. The samples were scanned from 2θ = 3° to 45° at the step scan mode, and the diffraction pattern was recorded using a scintillation counter detector. Raman spectra were collected in the range from 700 to 3000 cm−1 using Bruker Dispersive Raman Spectrometer fitted with a 785 nm laser source. The molecular weight of copolymer was estimated at a flow rate of 1.0 mL min−1 at 25 °C by gel permeation chromatography (GPC) on a Waters instrument (Waters Corporation, USA), using DMF as eluent, and the molecular weight was determined vs. polystyrene standards. X-ray photoelectron spectroscopy (XPS, PHI5000 ESCA, Perkin Elmer, USA) equipped with Al Kα source (1486.6 eV photons) was used to characterize the samples. The compositions of the GO–Au NPs hybrids catalysts were analyzed by 4300 DV inductively coupled plasma-atomic emission spectroscopy (ICP-AES, USA) on a Thermo Elemental IRIS Intrepid.3. Results and discussion
3.1 Characterization of Au NPs–GO hybrids
FTIR can reveal the characteristic vibrations of P(OEGMA-co-MQ) copolymer brushes in the obtained GO hybrid after polymerization. As shown in Fig. 1, the main typical absorption bands of GO at 3433 (O–H), 1726 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O carbonyl stretching of COOH) and 1000–1410 cm−1 (C–O in C–OH/C–O–C) can be observed.29 Among them, the resonance at 1621 cm−1 could be attributed to the vibration of the adsorbed water molecules, but may also contain the skeletal vibrations of unoxidized graphitic domains.15 The above absorption bands indicate that the abundant oxygen-containing functional groups are present on the GO surface. The esterification condensation between GO and RAFT agent results in the appearance of new bands at 1646, 1387, 1214, 1100 and 807 cm−1 in the curve of GO-RAFT (Fig. 1b). Herein, the characteristic peak at 1646 cm−1 is ascribed to the existence of the carbonyl stretching vibration of the esters, and the others are related to the incorporation of the RAFT agent on the surface of GO.15 After the RAFT polymerization, an obvious enhancement of the peak at 1646 cm−1 is observed in the curve (Fig. 1c) of the copolymer brushes–GO hybrids. Such observation results from the copolymerization of MQ and OEGMA on GO, which induce much more ester carbonyl groups on the surface of GO. Besides, a new peak appears at 1731 cm−1, which can be ascribed to the CO vibration of MQ.27 As expected, the FTIR data demonstrate the covalent attachment of the copolymer chains of P(OEGMA-co-MQ) to the GO sheets.
O carbonyl stretching of COOH) and 1000–1410 cm−1 (C–O in C–OH/C–O–C) can be observed.29 Among them, the resonance at 1621 cm−1 could be attributed to the vibration of the adsorbed water molecules, but may also contain the skeletal vibrations of unoxidized graphitic domains.15 The above absorption bands indicate that the abundant oxygen-containing functional groups are present on the GO surface. The esterification condensation between GO and RAFT agent results in the appearance of new bands at 1646, 1387, 1214, 1100 and 807 cm−1 in the curve of GO-RAFT (Fig. 1b). Herein, the characteristic peak at 1646 cm−1 is ascribed to the existence of the carbonyl stretching vibration of the esters, and the others are related to the incorporation of the RAFT agent on the surface of GO.15 After the RAFT polymerization, an obvious enhancement of the peak at 1646 cm−1 is observed in the curve (Fig. 1c) of the copolymer brushes–GO hybrids. Such observation results from the copolymerization of MQ and OEGMA on GO, which induce much more ester carbonyl groups on the surface of GO. Besides, a new peak appears at 1731 cm−1, which can be ascribed to the CO vibration of MQ.27 As expected, the FTIR data demonstrate the covalent attachment of the copolymer chains of P(OEGMA-co-MQ) to the GO sheets.
 | ||
| Fig. 1 FTIR spectra of GO (a), GO-RAFT (b) and copolymer brushes–GO (c). | ||
The significant structural changes of the materials from GO to copolymer brushes–GO hybrid during the modification process have been demonstrated by Raman spectra. As presented in Fig. 2, the curve of GO shows two prominent well-documented peaks at around 1342 cm−1 (D band) and 1596 cm−1 (G band). The appearance of D band at 1342 cm−1 is resulted from the vibration of carbon atoms of disordered graphite, indicating the formation of sp3 carbon in GO. Whereas the appearance of G band at 1596 cm−1 is associated with the first order scattering of E2g vibration mode for sp2 carbon lattice of graphitic domain.28–31 After functionalized with the RAFT agent and following copolymerization, the GO-RAFT and copolymer brushes–GO hybrid show similar peak positions of D band and G band with GO (see Fig. 2b and c). But the intensity ratio of ID/IG increases from 0.96 (GO) to 0.98 (GO-RAFT) and 1.13 (hybrid). It is quite obvious that the intensity ratio of GO-RAFT (ID/IG = 0.98) is similar with that of GO (ID/IG = 0.92). Such observation could be ascribed to the change of sp2 domains is accompanying with the change in the average size of sp3 domains upon GO, which just counteract each other out.31 As compared with GO, the ID/IG ratio of copolymer brushes–GO hybrid enhances to 1.13 due to the increasingly disordered structures, indicating the gradual increase of sp3 carbon structure after copolymerization.30 Furthermore, we can calculate the size of sp2 carbon clusters of GO and copolymer brushes–GO hybrid by the Knights empirical formula:32
| La = 4.35/(ID/IG) | (1) |
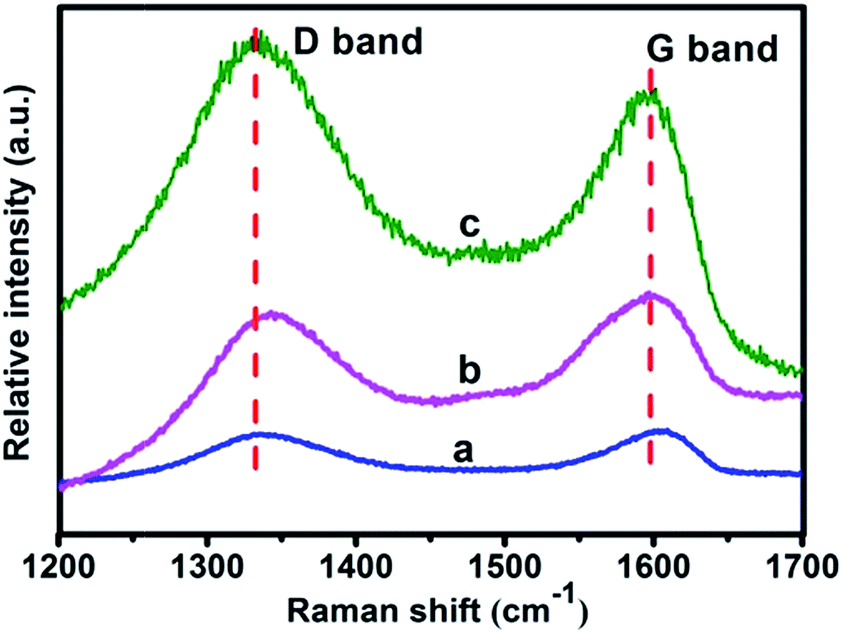 | ||
| Fig. 2 Raman spectra of GO (a), GO-RAFT (b) and copolymer brushes–GO (c). | ||
The structures of graphite, GO and copolymer brushes–GO hybrid were investigated by power XRD patterns (see Fig. 3). For graphite, the strong and sharp characteristic peak at 2θ = 26.5° indicates a higher ordered structure corresponding to a basal spacing d002 = 0.334 nm.33 After the oxidation of the graphite (Fig. 3b), the representative diffraction peak (001) of GO is observed at 2θ = 11.01° with a corresponding interlayer spacing (d) of 0.80 nm.31 As compared with the basal spacing of graphite (∼0.334 nm), the distinct increasing of the interlayer distance can be attributed to the presence of oxygen-containing functional groups.33 When the P(OEGMA-co-MQ) copolymer is modified onto GO, two broad diffraction peaks appear at 10.23° and 23.05°. The diffraction peak at 10.23° is associated to the inter-molecular backbone–backbone correlation and the size of the side group which is originated from the approximately hexagonal ordering of the molecular chains.31 The broad peak at 23.5° indicates that the nanosheets have poor order along the stacking direction and many free nanosheets are existed in the sample.34
 | ||
| Fig. 3 XRD patterns of graphite (a), GO (b) and copolymer brushes–GO (c). | ||
The TEM images of GO, copolymer brushes–GO hybrid and Au NPs–GO hybrid-1 are shown in Fig. 4. For the pristine GO, many of wrinkles and folding are displayed in the GO sheets, which seems as an exfoliated crumpled thin flake.29,35 When a comparison is made between the pristine GO and the copolymer brushes–GO hybrid (Fig. 4b), we can clearly observe that the resulting copolymer brushes–GO hybrid presents the thin homogeneous polymer layer on the surface of GO with the dark area over the GO sheets. The result implies the formation of copolymer brushes from the GO surface.29 The TEM and HRTEM images collected from the GO–Au NPs hybrid-1 are shown in Fig. 4c and d. It is obvious that the copolymer brushes–GO hybrid has been decorated with many small Au NPs and the average diameter of the Au NPs is about several nanometers (<10 nm). The well-resolved lattice planes of 0.24 nm spacing in the HRTEM image testify the crystalline structure of Au NPs formed on the copolymer brushes–GO. In addition, the Au NPs were enwrapped by the polymer brushes with 8-hydroxyquinoline ligands on the side chains via the coordination interaction in the dried state for the sample preparation of TEM testing. Actually, the Au NPs attached copolymer brushes–GO should exhibit a more extended chain conformation for the water-soluble polymer brushes in water, which is beneficial to improve the catalytic efficiency. The TEM and HRTEM images of Au NPs–GO hybrid-2 are shown in Fig. S1.† It is distinct that the copolymer brushes–GO hybrid has also been decorated with many Au NPs and the HRTEM image testifies the crystalline structure of Au NPs formed on the copolymer brushes–GO. However, the average diameter of Au NPs in the Au NPs–GO hybrid-2 is much larger than that of Au NPs–GO hybrid-1 (>20 nm).
 | ||
| Fig. 4 TEM images of GO (a), copolymer brush–GO (b), Au NPs–GO (c) and HRTEM image of Au NPs–GO hybrid-1 (d). | ||
XPS measurements were performed to reveal the valence states of GO, GO-RAFT, copolymer brushes–GO and Au NPs–GO hybrid-1. As shown in Fig. 5, when compared with the pristine GO, there is an obvious decrease in the O:C ratio along with a new peak at 166.8 eV corresponding to the binding energy of S 2p in the GO-RAFT. The result could be attributed to the introduction of the RAFT agent with trithiocarbonate groups to the GO sheets. After the introduction of P(OEGMA-co-MQ) chains on GO, a new peak at 397.1 eV which assigned to the N–H of the MQ segments of the P(OEGMA-co-MQ) chains appears in the copolymer brushes–GO hybrid. However, the binding energy of S 2p hardly be observed in the copolymer brushes–GO hybrid, because the content of the element S is too little in the copolymer brushes–GO. As expected, the evident existence of the element N in the copolymer brushes–GO provides additional evidence for successful polymerization. The high-resolution XPS Au spectrum of Au NPs–GO hybrid-1 is shown in Fig. 6. The binding energy (BE) for Au 4f7/2 of the Au NPs is 84.2 eV, which falls midway between Au(0) (BE = 83.9 eV) and Au(I) (BE = 85.1 eV), suggesting the coexistence of Au(I) and Au(0) in the Au NPs.36 The XPS signature of Au 4f doublet (4f7/2 and 4f5/2) is deconvoluted into two pairs of peaks (84.2 and 83.1 eV), corresponding to the reduced Au(0) clusters and the Au(I) ions, respectively. From the ratio of areas between the above peaks, we can calculate that approximately 44% of Au is on the surface of GO–Au NPs hybrid-1 as Au(I) which is coordinated with the 8-hydroxyquinoline ligands of copolymer brushes on GO.
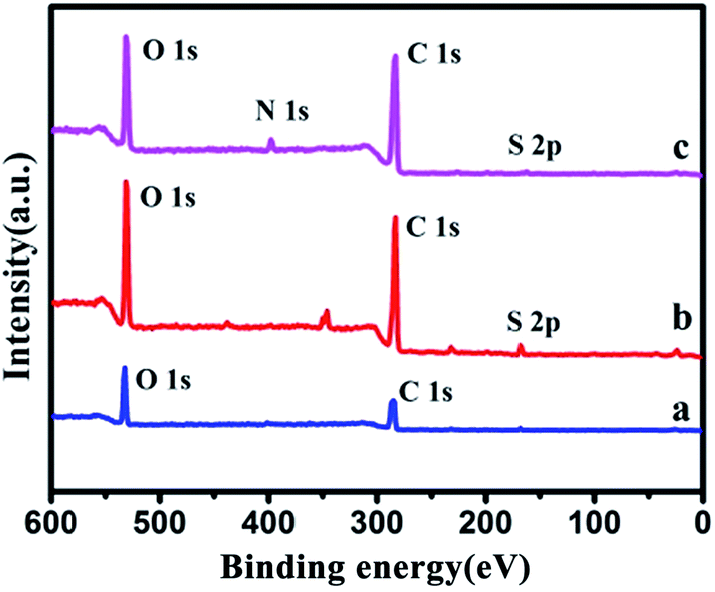 | ||
| Fig. 5 XPS spectra of GO (a), GO-RAFT (b) and copolymer brushes–GO (c). | ||
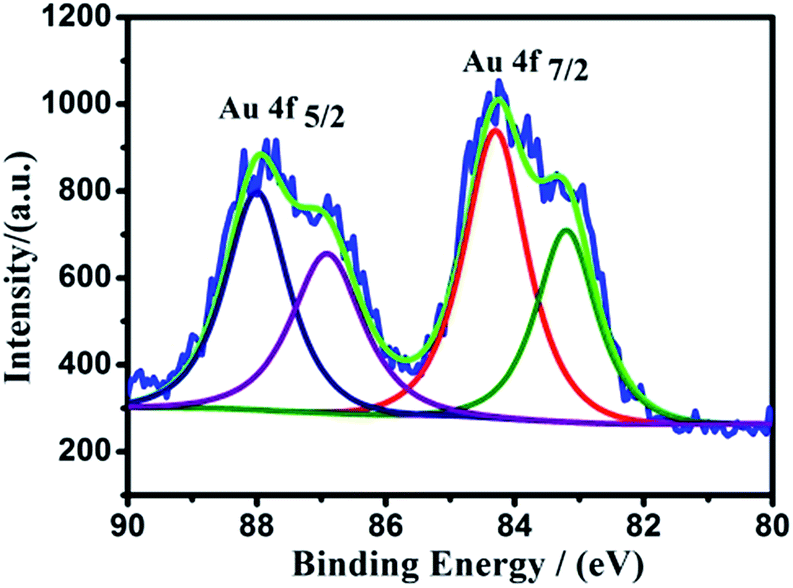 | ||
| Fig. 6 XPS spectra of Au NPs–GO hybrid-1. | ||
The evidence for the thermal stability of GO and copolymer brushes–GO hybrid could be obtained from TGA measurement. As shown in Fig. 7a, the slightly small mass loss (8 wt%) of GO below 100 °C is corresponding to the moisture contents of GO.37 The second acute weight loss (30 wt%) of GO takes place around 200 °C resulted from the pyrolysis of labile oxygen functional groups.37 Then, the weight loss of GO is much slower between 220 and 700 °C. The total weight loss of GO is 45 wt% (except water). For the copolymer brushes–GO hybrid (see Fig. 7b), the weight loss below 200 °C decreases to some extent because that the copolymer brushes are much more stable than the abundant oxygen-containing functional groups of pure GO. The weight loss above 300 °C is mainly assigned to the decomposition of copolymer brush. Estimated from the TGA curve of GO and copolymer brushes–GO hybrid, the weight loss caused by copolymer brushes decomposition is about 38 wt%. From the TGA curves of pure GO and free copolymer (see Fig. 7c), it is instinct that the thermal stability of free copolymer is higher than that of GO, so we have reasons to conclude that the grafted P(OEGMA-co-MQ) brushes appear to be effective in enhancing thermal stability of GO. Furthermore, the TGA curve of copolymer brushes–GO hybrid further proves the above conclusion.
 | ||
| Fig. 7 TGA curves of GO (a), copolymer brush–GO (b) and free copolymer P(OEGMA-co-MQ) (c). | ||
The grafting density can be expressed in the following formula:38
Chains per carbon:
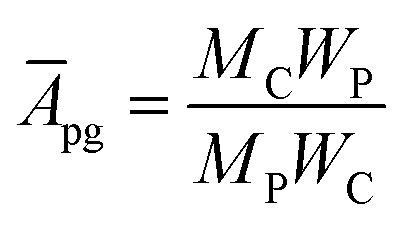 | (2) |
000 g mol−1. Then it can be calculated that the grafting density of the P(OEGMA-co-MQ) chains on GO sheets is 0.1 chains per 100 carbons.
3.2 Optical properties of Au NPs–GO hybrid
The UV-vis absorption spectra of GO, copolymer brushes–GO and Au NPs–GO hybrid-1 are depicted in Fig. 8. As shown in Fig. 8a, two typical characteristic absorption of GO can be observed at 230 and 300 nm. The maximum absorption at 230 nm results from the π–π* transition of aromatic CC bond, while the other absorption at 300 nm can be linked to the n–π* transition of CO bonds.39 After RAFT copolymerization (see Fig. 8b), a new absorption signal appears at 259 nm which is associated with the π–π* electron transition from quinoline ring.40 For the spectrum of GO–Au NPs hybrid-1 (Fig. 8c), the colloidal gold surface plasmon resonance band at around 520 nm is hardly observed, indicating that the Au NPs on the copolymer brushes modified GO should be very small. This result is consistent with the TEM image of GO–Au NPs hybrid-1. Therefore, the above results further evidence that the copolymer brushes have been grafted onto GO sheets and subsequent in situ synthesis of Au NPs on the GO surface has been carried out successfully.
 | ||
| Fig. 8 UV-vis spectra of GO (a), copolymer brushes–GO (b) and Au NPs–GO hybrid-1 (c). | ||
3.3 Catalytic behavior of Au NPs–GO hybrid
When the samples of copolymer brushes–GO were dispersed in aqueous solution, a homogeneous stable dark solution was observed at ambient temperature in the absence of any specific dispersant. More remarkably, the resulting dispersion could remain stable, and no obvious precipitation occurred even after storage under ambient conditions for more than seven days. It is interesting that the Au NPs–GO hybrid-1 in aqueous solution also exhibits weak fluorescent emission in the PL spectra (see Fig. 9). The copolymer brushes stabilized Au NPs on GO hybrid has a weak blue-light emission at 475 nm by adjusting pH values from 8 to 10 (λexc = 360 nm). The brightest emission is detected at pH = 9. To our knowledge, the fluorescence of Au NCs is originated from the gold core, and the increasing nanocluster size leads to lower energy emission. However, our present work has proved that the similar size-dependent tunable emission of Au NPs does not exist in our samples.40 Additionally, if the intrinsic properties of the Au8 cluster is the reason of the blue fluorescence, the relative size of Au NPs should be less than 1 nm. But as seen from our TEM images, even the smallest Au NPs are bigger than 1 nm. Therefore, we suggest that the origin of the weak blue emission of Au NPs–GO hybrid-1 should be ascribed to the ligand-to-metal charge transfer transition. As we all know, the luminescent Au NCs are expected to exhibit d and sp bands.40 In our sample, the most Au NPs are very small in the Au NPs–GO hybrid-1, so the energy level spacing within the sp band is too small to obtain strong fluorescent emission.40 Nevertheless, the electronic structures of metals are not merely dependent on their size, but they are obviously related to the oxidation state of their metal atoms.41 The 8-hydroxyquinoline units in the copolymer brush is an electron donor ligand and its p orbital is higher in energy than the d orbitals of Au(I), the overlapping of these orbitals leads to the formation of ligand charge transfer excited states. | ||
| Fig. 9 PL spectra of Au NPs–GO hybrid-1 at different pH from 8 to 10. | ||
The catalytic reduction of 4-NP to 4-AP by excess NaBH4 with the Au NPs–GO hybrid-1 as a catalyst at ambient temperature was monitored by measuring the time-dependent adsorption spectra of the reaction mixture solution. As shown in Fig. 10a, the strong absorption peak of 4-NP at 400 nm decreases quickly with the reaction time in the presence of Au NPs–GO hybrid-1 as catalyst, and completely diminishes when all the 4-NP is converted to 4-AP, while a new peak at 290 nm appears with time due to the conversion of 4-NP to 4-AP. In the other hand, we also researched the catalytic performance of the Au NPs–GO hybrid-2. As shown in Fig. S2,† the Au NPs–GO hybrid-2 could also as a catalyst to reduce 4-NP to 4-AP by excess NaBH4 at room temperature. But when the reaction time reaches 31 minutes, the conversion is only about 50%. As compared with the Au NPs–GO hybrid-2, the reduction system with Au NPs–GO hybrid-1 as catalyst needs only 6 minutes to achieve the full reduction of 4-NP by NaBH4. The instinct different catalytic activity between the above hybrids may be attributed to the different diameter of Au NPs in the copolymer brushes–GO. According to some reported literatures, smaller particles have higher catalytic activity than that of larger particles, resulting from the fact that smaller particles have a larger numbers of surface atoms available for catalysis, in other words, the higher rate of reduction involving smaller particles is due to the higher surface area. The observed rate dependence, in relation to particle size, could be attributed to a higher reactivity of the coordinatively unsaturated surface atoms in small particles as compared to low-index surface atoms prevalent in larger particles.42,43
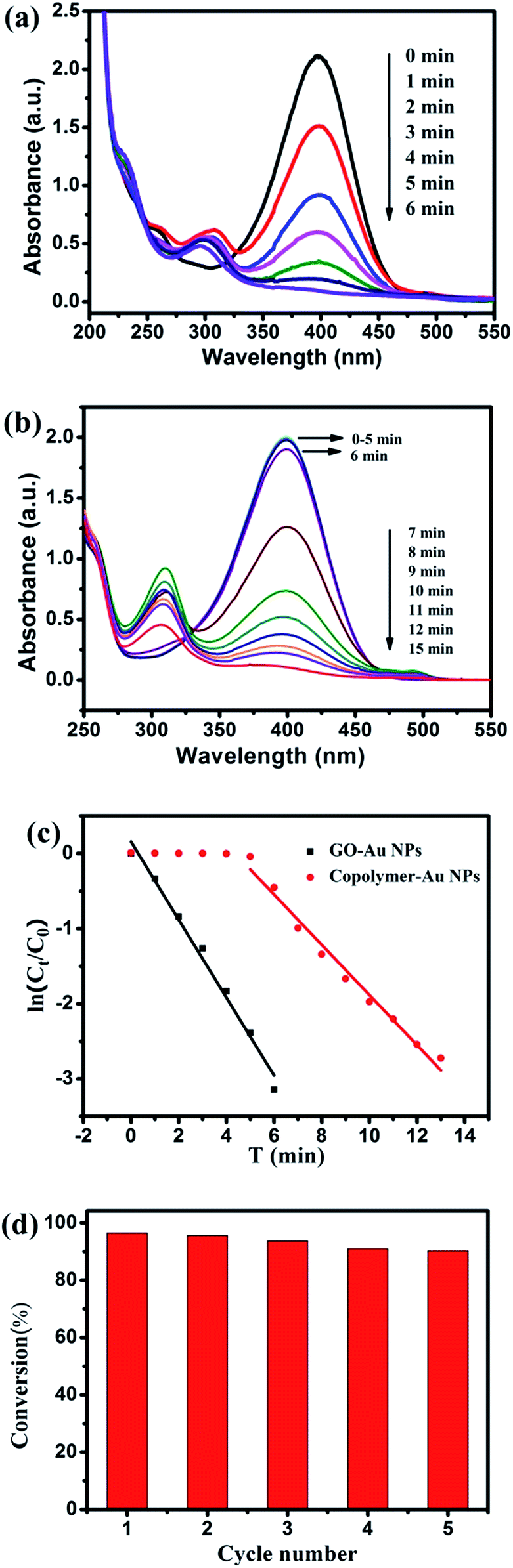 | ||
| Fig. 10 UV-vis adsorption spectra of the reduction of 4-NP by NaBH4 in the presence of Au NPs–GO hybrid-1 (a) and copolymer–Au NPs (b) and plots of ln(ct/c0) of 4-NP against time for the catalysts (c) and the conversion of 4-NP in five successive cycles with Au NPs–GO hybrid-1 catalyst (d). | ||
Because of the presence of large excess of NaBH4 compared to 4-NP, the rate of reduction is irrelevant of the concentration of NaBH4, and the reaction could be considered pseudo-first-order with respect to the concentration of 4-NP.44 The kinetic rate r is often defined:45
 | (3) |
 | (4) |
In our reduction process, ln(ct/c0) versus time can be obtained based on the absorbance as the function of time, and a good linear relationship can be observed (see Fig. 10c), which further indicates that the reaction follows pseudo-first-order kinetics. From the above formula (3) and (4), the kinetic reaction rate constant can be calculated at 0.52 min−1. Considering different conditions and different Au amounts for different catalyst systems, we used the turnover frequency (TOF) to determine the efficiency of Au NPs–GO catalyst for 4-nitrophenol reduction and compared the results with previously reported Au based nanocatalysts based on gold amount. The TOF value of Au NPs–GO hybrid-1 catalyst is 1.11 min−1, which value is calculated by the moles of 4-NP reduced per mole of Au per consumed time under the present reaction conditions. As shown in Table S1,† when compared with other Au based catalysts, our Au NPs–GO hybrid-1 catalyst shows a higher or similar catalytic efficiency than most of other Au based catalysts for the reduction of 4-nitrophenol.
In order to investigate the effects of the GO support on the catalytic activity, we also monitored the catalytic reduction of 4-NP to 4-AP with the copolymer P(OEGMA-co-MQ) stabilized Au nanoparticles as a catalyst at the same environment. As shown in Fig. 10b, the first 6 minutes in the catalyze process of copolymer–Au NPs is the induction time. The induction times have been attributed to many factors: (i) the diffusion-controlled adsorption of reactants onto the surface of the metal catalyst; (ii) the presence of dissolved oxygen that competes with 4-NP for NaBH4; and (iii) slow restructuring of the surface atoms of the nanoparticles.46–48 From Fig. 10b we know that a reaction time of 15 minutes is required to achieve the full reduction of 4-NP by NaBH4 using P(OEGMA-co-MQ) stabilized Au NPs alone as catalyst. Although a good linear relationship between ln(ct/c0) versus time can be observed in Fig. 10c, the kinetic reaction rate constants can be calculated to be only 0.33 min−1, suggesting that Au NPs–GO hybrid-1 has a better catalytic efficiency for the 4-NP as compared with P(OEGMA-co-MQ) stabilized Au NPs. We have made a comparison of catalytic activity between the two samples of copolymer–Au NPs and Au NPs–GO hybrid-1 in the similar analytical condition (see Fig. 10). The result shows that the Au NPs–GO hybrid-1 possess superior high catalytic activity than that of copolymer–Au NPs.4,21,49–52 Such observation indicate that the GO support may play an active part in the catalysis, which should be ascribed to the synergistic effect of GO nanosheet and Au NPs. The result could be explained as follows: 4-NP is π-rich in itself, the characteristic gives 4-NP an opportunity to be adsorbed onto GO via π–π stacking interaction. Such interaction provides more 4-NP molecules close to the Au NPs on the copolymer brushes–GO hybrid, leading to much higher probability for Au NPs to touch the 4-NP molecules.4,21 For the earlier studies, the catalysts and/or the catalyst supports are usually present in the form of particles, the adsorption of 4-NP on the surface of catalyst support is not significant like that on the surface of GO.50–52 The 4-NP molecules must collide with Au particles by chance, and remains in contact for the catalysis to proceed. Whereas if the collision is not accomplished, the 4-NP molecules will pass back into solution and can only react further when it collides with Au NPs again, the process merited lead to a slower reaction rate.21 On the other hand, the narrow size distribution, uniform distribution and good crystal structure of Au NPs for the Au NPs–GO hybrid-1 are beneficial to catalyze the reaction efficiently. What's more, there exists the electron transfer between GO and Au NPs, which can increase the local electron concentration and facilitate the uptake of electrons by 4-NP molecules.4,50–52
Additionally, the reusability of the Au NPs–GO hybrid as a heterogeneous catalyst was investigated. As shown in Fig. 10d, the Au NPs–GO hybrid was reused for five cycles for the reduction reaction of 4-NP with a very little catalytic loss, indicating the excellent stability of the obtained catalysts, which is attributed to the superior stability and dispersibility of Au NPs on the surface of GO sheets. All the results endow the possibility of the Au NPs–GO hybrid practical applications.
4. Conclusions
In summary, the water-soluble copolymer brushes grafted from GO were successfully fabricated via RAFT polymerization by using 8-hydroxyquinoline-containing monomer as ligands. It demonstrated that our strategy provided a simple and in situ reduction route for the synthesis of Au NPs on the copolymer brushes modified GO nanoplatform. The as-prepared Au NPs–GO nanomaterials exhibited a weak blue-light emission and good catalytic activity for the reduction of 4-NP, and the GO sheets could enhance the catalytic activity via a synergistic effect. Hence, considering the wide potential applications of the two-dimensional carbon material as a host material for a variety of nanoparticles, the approach developed here may lead to a new possibility for integrating active nanoparticles with graphene oxide nanosheets for catalysis, environment, and new energy fields.Acknowledgements
We would like to appreciate the financial support of the National Natural Science Foundation of China (21574017).Notes and references
- J. Huang, L. M. Zhang, B. Chen, N. Ji, H. F. Chen, Y. Zhang and Z. J. Zhang, Nanoscale, 2010, 2, 2733 RSC.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, M. I. Katsnelson, I. V. Grigorieva and S. V. Dubonos, Nature, 2005, 438, 197 CrossRef CAS PubMed.
- Y. Z. Zhou, X. Huang, X. Y. Qi, S. X. Wu, C. Xue, F. Y. C. Boey, Q. Y. Yan, P. Chen and H. J. Zhang, J. Phys. Chem. C, 2009, 113, 10842 Search PubMed.
- Y. Choi, H. S. Bae, E. Y. Seo, S. W. Jang, K. H. Park and B. S. Kim, J. Mater. Chem., 2011, 21, 15431 RSC.
- R. K. Layek and A. K. Nandi, Polymer, 2013, 54, 5087 CrossRef CAS.
- H. J. Salavagione, M. A. Gómez and G. Martínez, Macromolecules, 2009, 42, 6331 CrossRef CAS.
- Z. Liu, J. T. Robinson, S. M. Sun and H. G. Dai, J. Am. Chem. Soc., 2008, 130, 10876 CrossRef CAS PubMed.
- Y. Z. Pan, H. Q. Bao, N. J. Sahoo, T. F Wu and L. Li, Adv. Funct. Mater., 2011, 21, 2754 CrossRef CAS.
- H. Yang, K. Paek and B. J. Kim, Nanoscale, 2013, 5, 5720 RSC.
- H. K. He and C. Gao, Chem. Mater., 2010, 22, 5054 CrossRef CAS.
- D. Vuluga, J. M. Thomassin, I. Molenberg, I. Huynen, B. Gilbert and C. Detrembleur, Chem. Commun., 2011, 47, 2544 RSC.
- P. P Zhang, K. Jiang, C. N. Ye and Y. L. Zhao, Chem. Commun., 2011, 47, 9504 RSC.
- Y. Deng, Y. J. Li, J. Dai, M. D. Lang and X. Y. Huang, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1582 CrossRef CAS.
- M. Fang, K. J. Wang, H. B. Y. L. Lu and S. J. Yang, J. Mater. Chem., 2009, 19, 7098 RSC.
- B. Zhang, Y. Chen, L. Q. Xu, L. G. Zeng, Y. He, E. Y. Kang and J. J. Zhang, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 2043 CrossRef CAS.
- H. D. Huang, C. Y. Liu, D. Li, Y. H. Chen, J. G. Zhong and Z. M. Li, J. Mater. Chem. A, 2014, 2, 15853 CAS.
- K. Paek, H. Yang, J. Lee, J. Park and B. J. Kim, ACS Nano, 2014, 8, 2848 CrossRef CAS PubMed.
- C. Z. Zhu, L. Han, P. Hu and S. J. Dong, Nanoscale, 2012, 4, 1641 RSC.
- A. L. Pyayt, B. Wiley, Y. Xia, A. Chen and L. Dalton, Nanotechnology, 2008, 3, 660 CAS.
- S. J. Xu, L. Yong and P. Y. Wu, ACS Appl. Mater. Interfaces, 2013, 5, 654 CAS.
- Y. W. Zhang, S. Liu, W. B. Lu, L. Wang, J. Q. Tian and X. P. Sun, Catal. Sci. Technol., 2011, 1, 1142 CAS.
- C. J. Murphy, A. M. Gole, S. E. Hunyadi, J. W. Stone, P. N. Sisco, A. Alkilany, B. E. Kinard and P. Hankins, Chem. Commun., 2008, 5, 544 RSC.
- Y. W. Cao, R. C. Jin and C. Mirkin, Science, 2002, 297, 1536 CrossRef CAS PubMed.
- H. W. Hu, J. W. Xin, H. Hu, S. W. Wang, D. G. Miao and Y. J. Liu, J. Mater. Chem. A, 2015, 3, 11157 CAS.
- E. Lam and J. T. Luong, ACS Catal., 2014, 4, 3393 CrossRef CAS.
- C. J. Ferguson, Y. J. Hughes, D. Nguyen, B. Pham, R. Gilbert, A. C. H. Serelis and B. Hawkett, Macromolecules, 2005, 38, 2191 CrossRef CAS.
- N. Y. Du, R. Y. Tian, J. B. Peng and M. J. Lu, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 397 CrossRef CAS.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- A. Kundu, S. Nandi, P. Das and A. K. Nandi, ACS Appl. Mater. Interfaces, 2015, 7, 3512 CAS.
- K. Jiang, C. Ye, P. Zhang, X. Wang and Y. Zhao, Macromolecules, 2012, 45, 1346 CrossRef CAS.
- P. Ding, J. Zhang, N. Song, S. J. Tang, Y. M Liu and L. Y. Shi, Composites, Part A, 2015, 69, 186 CrossRef CAS.
- D. S. Knight and W. B. White, J. Mater. Res., 1989, 4, 385 CrossRef CAS.
- Q. Zhang, Q. L. Li, S. Xiang, Y. Wang, C. Wang, W. Jiang, H. Zhou, Y. W. Yang and J. Tang, Polymer, 2014, 55, 6044 CrossRef CAS.
- W. Z. Yuan, J. J. Wang, T. X. Shen and J. Ren, Mater. Lett., 2013, 107, 243 CrossRef CAS.
- W. F. Li, J. Y. Liang, W. T. Yang and J. P. Deng, ACS Appl. Mater. Interfaces, 2014, 6, 9790 CAS.
- C. Zhou, C. Sun, M. Yu, Y. Qin, J. Wang, M. Kim and J. J. Zheng, J. Phys. Chem. C, 2010, 114, 7727 CAS.
- Y. G. Shi, M. Y. Liu, K. Wang, F. J. Deng, Q. Wan, Q. Huang, L. H. Fu, Y. X. Zhang and X. Wei, Polym. Chem., 2015, 6, 5876 RSC.
- Y. S. Ye, Y. N. Chen, J. S. Wang, J. Rick, Y. J. Huang, F. C. Chang and B. J. Hwang, Chem. Mater., 2012, 24, 2987 CrossRef CAS.
- D. Li, M. B. Müller, S. Gilje, R. B. Kaner and G. G. Wallace, Nat. Nanotechnol., 2008, 3, 101 CrossRef CAS PubMed.
- B. X. Liu, Y. Wang, M. X. Deng, J. H. Lü, C. Y. Tong and C. L. Lü, RSC Adv., 2014, 4, 57245 RSC.
- J. Zheng, C. Zhou, M. X. Yu and J. B. Liu, Nanoscale, 2012, 4, 4073 RSC.
- S. Panigrahi, S. Basu, S. Praharaj, S. Pande, S. Jana, A. Pal, S. K. Ghosh and T. Pal, J. Phys. Chem. C, 2007, 111, 4596 CAS.
- N. T. Khoa, S. W. Kim, D. H. Yoo, E. J. Kim and S. H. Hahn, Appl. Catal., A, 2014, 469, 159 CrossRef CAS.
- F. H. Lin and R. A. Dong, J. Phys. Chem. C, 2011, 115, 6591 CAS.
- T. Wu, L. Zhang, J. P. Gao, Y. Liu, C. J. Gao and J. Yan, J. Mater. Chem. A, 2013, 1, 7384 CAS.
- S. Wunder, F. Polzer, Y. Lu, Y. Mei and M. Ballauff, J. Phys. Chem. C, 2010, 114, 8814 CAS.
- K. Kuroda, T. Ishida and M. Haruta, J. Mol. Catal. A: Chem., 2009, 298, 7 CrossRef CAS.
- Y. Mei, Y. Lu, F. Polzer and M. Ballauff, Chem. Mater., 2007, 19, 1062 CrossRef CAS.
- H. W. Hu, J. H. Xin, H. Hu and X. W. Wang, Nano Res., 2015, 8, 3992 CrossRef CAS.
- J. Luo, N. Zhang, R. Liu and X. Y. Liu, RSC Adv., 2014, 4, 64816 RSC.
- J. Chen, P. Xiao, J. C. Gu, Y. J. Huang, J. W. Zhang, W. Q. Wang and T. Chen, RSC Adv., 2014, 4, 44480 RSC.
- M. M. Zhang, X. Lu, H. Y. Wang, X. L. Liu, Y. J. Qin, P. Zhang and Z. X. Guo, RSC Adv., 2016, 6, 35945 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: TEM images, UV-vis and comparison of catalytic activity. See DOI: 10.1039/c6ra11710d |
| This journal is © The Royal Society of Chemistry 2016 |